Qualifying your cleanroom
Technology, manufacturing processes, and regulatory trends have changed in the 10 years since the last revision to Annex 1 of the EU GMP Guideline for the Manufacture of Sterile Medicinal Products. With the revised document set for release this year, the updates significantly impact quality control (QC), quality assurance (QA), and all laboratory activities. This revision is intended to add detail and clarity and provide global alignment of standards. Even though Annex 1 is a European document, drugs manufactured globally for sale in the EU need to comply with the standards, which is why Annex 1 was developed in collaboration with global regulatory bodies such as the World Health Organization and Pharmaceutical Inspection Co-operation Scheme.
Moreover, with the current economic boom for pharmaceuticals, more companies are expanding their manufacturing capabilities and opening new facilities. This is particularly relevant as, according to a survey from the Parenteral Drug Association, industry members find the most challenging proposed change in Annex 1 to be the qualification of facilities.
Constructing and qualifying a new cleanroom is a huge investment of a company’s money, time, and resources. Given the purpose of the cleanroom and its role in product quality, there are extensive regulatory requirements surrounding cleanroom design and validation. With recent revisions to some of these requirements in ISO 14644 and proposed changes to Annex 1, will your qualification meet the new standard?
Maintaining compliance
A recent U.S. Food and Drug Administration warning letter highlighted the potential consequences of failing to comply with GMP standards. In the letter, the FDA made the following observations regarding facilities and contamination control:
Drug products intended or expected to be sterile were prepared, packed, or held under insanitary conditions, whereby they may have become contaminated with filth or rendered injurious to health, causing your drug products to be adulterated under section 501(a)(2)(A) of the FDCA.
Your firm failed to establish and follow appropriate written procedures that are designed to prevent microbiological contamination of drug products purporting to be sterile, and that include validation of all aseptic and sterilization processes (21 CFR 211.113(b)).
Your firm failed to maintain the buildings used in the manufacture, processing, packing, or holding of a drug product in a clean and sanitary condition (21 CFR 211.56(a)).
Your firm failed to establish an adequate system for monitoring environmental conditions in aseptic processing areas (21 CFR 211.42(c)(10)(iv)).
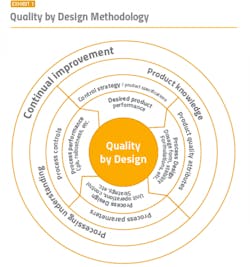
To maintain a strong compliance position, it is imperative to start with a solid foundation. A well-functioning cleanroom starts with good process and cleanroom design and utilizes key aspects of the well-established quality by design (QbD) methodology (Exhibit 1) and Quality Risk Management (QRM). The overall contamination control strategy should already be established and incorporated into the design. The appropriate layout, space, equipment, building materials, and facilities need to be determined based on the type of product and manufacturing cycles.
The flow of personnel and materials are critical aspects that also require special consideration. Annex 1, 2017 draft version section 4.2 states “the maximum number of operators in critical areas should be determined based on QRM, documented in the contamination control strategy, and validated during activities such as initial qualification and aseptic process simulations, so as not to compromise sterility assurance.” It is critical to control people and materials entering the classified clean areas.
The role of the QC microbiologist
The cleanroom design and qualifications should be overseen by a team representing (but not limited to) Engineering, Operations, Quality, Facilities and Microbiology. The Annex 1 draft from 2017 emphasizes the role of the QC microbiologist for all manufacturing processes. Specifically, in terms of cleanroom qualifications, it states in 2.c that “processes and monitoring systems for sterile product manufacture must be designed, commissioned, qualified and monitored by personnel with appropriate process, engineering and microbiological knowledge.”
It is important to recognize the many tasks of the QC microbiologist and their responsibilities during the planning and setting up a new cleanroom. While QC microbiologists are integral to the manufacturing design and processes, they are also the primary owner of the microbiology laboratory. Their overall list of responsibilities is comprehensive and might look similar to this:
- Design the laboratory and workflow
- Order, install and qualify equipment
- Hire and train technicians
- Write and revise SOPs
- Develop an environmental monitoring program
- Establish sampling plans
- Implement and validate QC methods and testing based on release specifications
- Conduct bioburden and endotoxin testing
- Perform microbial identifications
- Investigate out of specifications, deviations and contamination events
- Manage LEAN activities and productivity projects
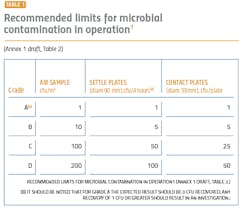
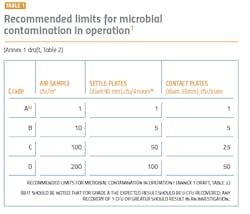
The performance qualification is when the surface and airborne particulate and bioburden levels are monitored and tested (see Tables 1 and 2). The qualification is absolutely essential for determining whether the cleanroom was adequately designed and built to prevent contamination of the products being manufactured and requires thorough microbiological analyses. A poor qualification increases risk to manufacturing, the environment, patient safety, and the company’s investment in the new facility.
Revised guidelines
The revised Annex 1 treats Class A (class 100/ISO 5) and Class B (class1000/ISO 7) equally in terms of monitoring and requalifications. These areas will need a higher number of sampling points for the aseptic processing and the immediately adjacent environment (grade A/B) (Annex 1, 2017 draft version: 5.26). Sampling points should include all critical processing locations, based on risk to the final product. The larger number of sampling points can contribute to a better understanding of the state of control and potential changes and trends.
During performance qualification, the bioburden counts need to be followed by microbial identification. Understanding the flora in the manufacturing environment serves to direct an appropriate cleaning strategy, helps determine where the contamination is coming from and what controls need to be added to protect the product. Risk assessments are an important part of qualifications, and an appropriate risk assessment cannot be made without an accurate identification.
The identifications generated during cleanroom performance qualification establish a baseline as to what flora is found in the manufacturing environment, and are also critical for establishing alert and action limits. From this information, changes in the manufacturing environment can be assessed.
According to Annex 1, 2017 draft version: 9.33, “If microorganisms are detected in a grade A or B zone, they should be identified to the species level and the impact of such microorganisms on product quality (for each batch implicated) and state of control should be evaluated. Consideration may also be given to the identification of grade C and D contaminants, and the requirements should be defined in the contamination control strategy.” It cannot be understated how important the accuracy and reproducibility of the microbial identifications are, especially for the initial qualification as it serves as a baseline.
Moreover, the cleanroom qualification is not a one-time event. Cleanrooms should be requalified biannually, as well as after changes to equipment, facilities or processes. It is clearly stated in the reviewed Annex 1 guideline (section 5.29) that for grades A/B the maximum time interval for requalification is six months. For grades C and D, the maximum time interval for requalification is 12 months.
The requalification gives the opportunity to critically evaluate the microbiological data. The microbial identifications are just as important during the requalification in order to assess changes and risk.
The draft Annex 1 also speaks extensively about the importance of trending data on a routine basis, not just during requalification. To adequately assess changes in flora type and numbers, there has to be complete confidence and reliability on the previously collected data. Frequent analysis of trends allows a proactive response to changes in the manufacturing environment. It helps identify controls to mitigate risk and protect the final product from contamination. It is not uncommon for regulatory inspectors to request trend reports and objective documentation for review.
Risks of a failed cleanroom qualification
If the microbial identification method is inaccurate or irreproducible during the performance qualification, there is a false sense of control, additional time and money wasted to remediate contamination (both at the time of the qualification and in the future), ineffective action and alert limits, and delays in manufacturing. Those delays may cause drug shortages which would have a direct impact on patients and draw unwanted attention by regulatory bodies.
Since a new facility is under severe scrutiny by company executives, these undesirable outcomes can be very difficult to explain to management.
Outsourcing microbial identifications
Given the extent of the QC microbiologist’s job functions and importance, as well as the importance of microbial identifications to the cleanroom’s performance qualification, outsourcing identifications is a logical and beneficial option. Most commercial systems have limited throughput and cannot identify that large of a volume of microorganisms in a reasonable amount of time. All the data then must be compiled into a report and analyzed, which takes even more time and labor by the micro lab. Every day that it takes to complete the performance qualification means thousands of dollars in lost revenue.
The new Annex 1 draft also emphasizes the use of “scientifically sound and modern methods.” This applies also to microbial identifications. Phenotypic systems that identify organisms based on biochemical and metabolic properties have been around for decades. The libraries associated with these systems are often limited and focused on clinical isolates. The identification rates can be subpar, and the number of retests requires more time, resources and cost. Identifications with phenotypic systems are less accurate and reproducible, especially as manufacturing isolates are stressed from the nutrient-poor environment and biochemical characteristics may not be expressed.
On the other hand, modern identification methods include genotypic DNA sequencing and proteotypic identifications using a MALDI-TOF mass spectrometer. DNA sequencing remains the “gold standard” for identifications, but typically requires more labor, more equipment, and technical expertise. Proteotypic identification methods do not offer phylogenetic information but are fast and reproducible. The MALDI-TOF instrument is a significant investment, so many companies choose to outsource both DNA sequencing as well as MALDI-TOF identifications.
Considering that the importance of accurate and reliable microbial identifications is clearly emphasized for cleanroom qualifications in the Annex 1 draft, the optimal identification method is DNA sequencing.
When selecting an outsourcing lab partner, consider the availability of tracking and trending data tools. As mentioned in the previous section, Annex 1 expects frequent trending of environmental monitoring samples. Having trending organism reports included with the identification service could save significant time and reduce the risk of transcription errors. Thus, improving your data integrity position, and strengthening your compliance position. The trending reports can give a complete breakdown of what microorganisms were recovered from your cleanroom, allows you to easily view changes from your baseline, and assess remediation efforts.
Outsourcing your microbial identifications to a reputable company using robust and accurate services can give you confidence in your performance qualification. It can also speed up your timeline, allow you to make informed decisions about your process and contamination controls, decrease contamination risk, and save precious time and resources.
Another critical factor in selecting an outsourcing partner for microbial identifications should be appropriate lab accreditations such as ISO 17025. This accreditation certifies the technical competence of laboratories to perform specific tasks and guarantees that the results are obtained according to valid methods and procedures that comply with precise standards.

Worth the investment
The revised Annex 1 guideline places renewed emphasis on facilities and contamination control. This directly impacts cleanroom qualifications, which play a critical role in assessing the manufacturing state of control and establishing a baseline for assessing changes and risk to the final product. Accurate and reproducible microbial identifications in combination with meaningful tracking and trending of the generated results are necessary for effective contamination control and outsourcing these identifications can be beneficial.
Cleanroom qualifications are a huge investment of a company’s money, time and resources. A strong and comprehensive qualification increases the company’s regulatory compliance position, enhances product quality, and ensures patient safety.
References
1. ISO 14644-1:2015, “Cleanrooms and associated controlled environments — Part 1.”
2. Luke Christou, “The high cost of contamination in drug manufacturing,” Pharmaceutical Technology, Sept. 12, 2018.
3. FDA Warning Letter, March 20, 2019.
4. Damon Larkin, “The contamination risk posed by laundered cleanroom apparel,” Kimberly-Clark Professional, November 2014.
5. Edward Tidswell, “The cost of microbial control,” PDA Letter, Aug. 8, 2017.
6. David Westman, “Cost and Impact of a bioburden incident,” GE Healthcare Life Sciences April 12, 2017.