Today's solid dose drugs: High potency, high stakes
As oral solid dose (OSD) drugs in development become more potent, regulatory expectations keep changing. To sustain compliance proactively, drug developers and their partners must develop a clear understanding to navigate current complex regulatory environments and build their manufacturing strategies accordingly.
Otherwise, you or your manufacturing partners might be at risk, blindsided during a facility inspection with a finding that not only will slow your drug’s pace to market, but jeopardize its regulatory approval altogether.
Keeping up with market growth
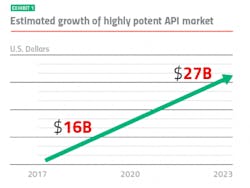
While regulators mandate protecting workers and preventing cross-contamination, historically they have never provided well-defined, harmonized standards for how to do it. It is up to manufacturers to determine how to meet regulators’ expectations and demonstrate to regulators their methods are sufficient.
Various sources have created charts, lists and formulas for determining the risks posed by these drugs, along with a variety of recommendations for how to manufacture them safely. But without consistent guidelines, each pharmaceutical company has had to develop its own system for meeting regulator expectations. To make it even more challenging, the regulatory landscape has been shifting as the manufacturers have been responding to the development and growth of these potent compounds.
Regulatory models for manufacturing
Today, the regulatory environment is simultaneously governed by two main models: the “traditional” model, which has been in play for decades; and a newer, health-based model originated by the European Medicine’s Agency (EMA). The health-based approach was adopted more recently by an international consortium of regulatory bodies, known as the Pharmaceutical Inspection Convention and Pharmaceutical Inspection Co-operation Scheme or PIC/S — whose stated mission is to harmonize cGMP standards around the world.
Similarities
Notably, the traditional and health-based models possess striking similarities. Regarding implantation, each has effective, and each has less-effective methodologies to attain compliant containment. The absolute goal of containment is to protect workers and the environment from ambient, environmental exposure to the highly potent, highly active dusts and powders involved in the manufacturing process. This control is also necessary to help eliminate cross-contamination.
As a first line of defense, regulatory authorities prefer engineering controls — containing the compound within the manufacturing equipment so it doesn’t escape into the work environment. Personal protection equipment, such as “bunny suits,” provides supplementary protection.
To keep workers safe, plant operators periodically monitor the air employees breathe during the production process in order to ensure it meets acceptable exposure limits for the drug they are manufacturing.
While various methods are used to determine these limits — commonly referred to as Occupational Exposure Limits (OELs) — they are mostly aligned and accepted within the traditional and health-based frameworks.
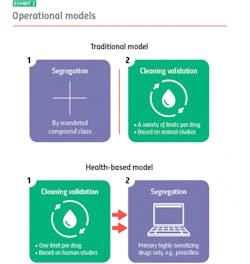
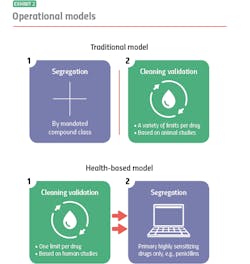
Where the two models diverge is their approach to segregation and cleaning validation, for example: With the traditional model, you segregate, and then you clean. With the health-based model, you clean, and if you can’t clean well enough, you segregate.
Segregation refers to manufacturing drugs in separate facilities or self-contained areas. Originally, segregation was intended to prevent the cross-contamination of highly potent or highly active drugs, though that has largely changed due to advances in cross-contamination control.
Cleaning validation on the other hand provides cross-contamination control at the equipment level. Its goal is to protect patients taking the next drug made in the same equipment. After manufacturing a drug, you clean your equipment, and then you validate how well you’ve cleaned it. Operations technicians do this usually by assessing samples taken from inside the equipment and/or of the liquid used to rinse out manufacturing equipment after it’s been cleaned. They can then identify if any remaining residue meets acceptable safety limits.
Traditional model: segregation then cleaning
The traditional model mandates segregation by designated compound class, primarily to meet compliance for those countries that require it. You manufacture hormones in one facility or segregated area, immunosuppressants in another, sensitizers in another and so on. Everything else you can manufacture together in a general tableting or general solids area using your own cross-contamination control strategy.
When it is time to clean, it is time to meet safety limits per drug derived — primarily from animal toxicology studies. Operators can choose from a variety of generally accepted measurements, including toxicity thresholds (TTC) and lethal-dose limits (LD50s) based on rat studies.
Under the traditional model, segregation and cleaning validation are separate, unrelated activities. Segregation is segregation, and cleaning validation is cleaning validation.
Health-based model: cleaning then segregation
The health-based model takes an entirely different approach. Under this model, segregation is dictated by how well operators can control cross-contamination, with cleaning validation (which are based on different criteria) playing a pivotal role. Here, it is best practice to clean to meet a single safety limit per drug derived primarily from human safety data from clinical trials.
If you’re able to meet that limit, you usually don’t have to segregate: You’ve cleaned your equipment so well that whatever residue might remain will not contaminate the next drug made in that equipment or harm the patients who take that drug. If you cannot achieve the safety limit, you segregate.
Regardless of model, health-based or traditional, cleaning validation is only one part of a comprehensive cross-contamination control strategy. If you can’t demonstrate contamination control for whatever reason, then it is time to segregate.
Currently, under the health-based model the only drugs that always need segregating, regardless of an organization’s cleaning prowess, are highly sensitizing compounds such as beta lactam antibiotics (i.e. penicillins). There is no acceptable exposure limit for these drugs because even imperceptible amounts may harm, or even kill people who are allergic.
Today’s reality
How do you manufacture highly potent drugs compliantly in this complex and evolving environment? It’s a challenge. While the industry is moving slowly toward the health-based model — more than 50 countries have formally signed on — the regulatory landscape remains in flux.
The reality on the ground is that inspectors who come to your facility may use either or both models to evaluate your operations: An inspector from a country that has accepted the health-based approach may still look for facilities segregated by traditional compound class. An inspector from a country using the traditional model may also want to see health-based elements in your manufacturing program. What’s a manufacturer to do?
Pfizer’s approach
Pfizer manufactures highly potent oral solid dosage forms at several facilities designed specifically for that capability. Given the regulatory scenario, Pfizer sought to create a safe, consistent and compliant approach to manufacturing that would serve Pfizer and Pfizer CentreOne’s customers long term.
The Newbridge Ireland facility produces high-potency tablets, capsules and pellets. Since these medications are marketed in more than 100 countries, Pfizer needs to meet the scrutiny of inspectors from a number of jurisdictions, regardless of the regulatory model they use.
To assure international compliance at Newbridge, Pfizer took a dual approach conforming infrastructure and processes to both the traditional and health-based models. Because the company operates across the regulatory spectrum, as the industry moves in the health-based direction, Pfizer and its contract customers move along with it while continuing to meet contemporary expectations.
A plant-in-plant concept
Pfizer built the Newbridge plant to meet the segregation requirements of the traditional approach. The company’s “plant-in-plant” design appears as one building from the outside, with a single roof covering almost a million square feet. However, lift off that roof and people will find 10 self-contained areas, each with its own air handling, material flow and personnel flow to achieve complete segregation.
The traditional model mandates segregation by specific compound class. Unfortunately, the point where to “slot” each compound varies among regulators around the world. At Newbridge, they use a Pfizer-proven process to classify each compound based on toxicological information derived from animal and human studies. The resulting classification dictates whether or not they should segregate.
For drugs that do not require segregation — and can be safely manufactured in a shared area — we conduct a formal quality risk assessment to make sure we have the technical and organizational controls in place to prevent cross-contamination.
This approach has served Pfizer well. As of this writing, the plant has been able to meet segregation requirements regardless of country throughout the facility’s history.
Cleaning validation
As with segregation, Pfizer designed its cleaning validation criteria to satisfy both regulatory models. As a whole, traditional cleaning limits tend to be the more conservative, providing a wide margin of safety to allow for the indirect animal-to-human translation of study data. On the other hand, health-based cleaning limits are sometimes more generous: They’re derived directly from human data so provide relatively fine-tuned margins of safety for human exposure.
Regulators, however, haven’t loosened their requirements for cleaning levels, regardless of model. To satisfy the most stringent expectations, we base our cleaning limits on both traditional and health-based calculations, and clean to the most conservative limit.
Containment
While getting segregation and cleaning validation right is critical, the most important health concern is safeguarding colleagues who handle these drugs every day. Even minimum exposure can have health consequences due to the potency of these compounds and frequency of contact.
To protect workers, the level of containment must be matched to a drug’s risk. But no formula exists to make that determination. To provide a starting point for decision-making, Pfizer developed “Exposure Control Practices.” Used across our global network, these procedures range from how to perform risk assessments to criteria for facility design, including room planning, air locks and exchange rates, and personal protective equipment.
While Pfizer provides criteria for equipment selection, facilities are not required to purchase specific models of tablet presses or fluid bed dryers, for example. At Newbridge, as at other Pfizer facilities, surveying the market and purchasing equipment that best matches the needs of projects and manufacturing environments maintains flexibility. This kind of flexibility enables Pfizer to stay current with technologies that are quickly evolving to better protect operators from exposure and control cross-contamination.
Finally, no amount of technology can safeguard the manufacturing environment without well-trained staff. Staff must understand how to handle these specialized compounds and systems — and themselves — with skill and compliance.
Keep up your commitment to compliance
As you manufacture highly potent compounds, in-house or through a CDMO, know that you need a manufacturing strategy that covers both the traditional and health-based regulatory models. Without it, you’re taking a risk each time an inspector arrives at your door.
You need to keep up with containment requirements; it’s not a static environment. Stay on the curve, if not ahead. And be committed. That’s the hardest part of manufacturing! To stay in compliance, you need resources: the right people, enough time, and sufficient money to invest over the long term.
References
1. HPAPI Market by Type. Markets and Markets. April 2018