Juggling the regulatory complexities of global clinical trials

Regulatory compliance continues to be a complex and cumbersome requirement for sponsors of clinical trials. These regulations require interpretation by experienced and qualified individuals who can ensure a thorough understanding of the requirements and how they can best be applied to drug development.
Ultimately, it is always the responsibility of the clinical trial’s sponsor to meet regulatory compliance and defend the data that is reported. Due to the complexity of the task, the effort is supported by a network of professionals with different backgrounds: clinical trial teams, regulatory affairs and pharmacovigilance. Deploying an automated technology process to safety and compliance information via a central hub is one way to simplify tracking country rules for compliance. This approach can feature dynamic templates, which can be modified to meet local requirements, help to set a standard for tracking each country’s regulations, and upon distribution to clinical trial investigators, can be blinded to avoid putting clinical trial information in jeopardy.
Distributing safety documents remains a very manual process today (even when using technology like a portal). New technology can automate this process by using the knowledge that is inherent to the core sets of data that drive distribution.
The safety letter itself has metadata about the originating study, compound, originating country etc. This helps to automate the process by combining safety letter metadata with a distribution list and meeting various country regulations.
Importantly, embedded country rules are checked with the distribution list to ensure only the required contacts are notified (this removes over-reporting and ensures regulatory compliance).
Automation removes significant manual efforts (which are both costly and error-prone) and speeds up the distribution process — meaning documents are distributed in real time with no human intervention required.
Cross-border regulation updates
Regulations are usually high-level, which means that general guidelines are provided but specific details and the processes needed to comply are not, leaving room for interpretation. Country-level guidelines then need to be translated into practical actions that can be applied across trial sites and institutions. In many countries, local competent authorities can apply their interpretations of these guidelines, creating complexities for clinical trial management teams when it comes to safety reporting. Additionally, local and central ethics committees often add their subtleties and fail to distribute notifications about these updates or changes. This causes even greater challenges for sponsors in maintaining up-to-date information on regulatory compliance and applying this information correctly during a trial.
Also, many countries publish their regulations in local languages. This requires knowledgeable interpreters who can grasp the nuances of the language and deliver accurate and transparent regulatory updates and alerts to clinical trial managers and sponsors.
Thus, for a sponsor evaluating a compound with clinical trials in various global geographies, management of regulatory compliance becomes increasingly convoluted. For example, if you are conducting a trial in Luxembourg for a drug that will be marketed across Europe and North America, you need to not only consider regulations that apply to the country trial site but also regulations in all countries where the drug will be available for distribution. Compliance with multiple countries’ requirements requires significant resource allocations and standardization of data capture and reporting, a problem that a centralized hub could help fix by facilitating away some of these data delivery to customers who only have to focus on just a ‘yes or no’ acceptance of the standards.
When countries don’t advise of regulatory modifications
Generally, in the U.S. and EU, there is notice when there’s a change of law. However, this is not the global standard. In compliance, there can be “oops” moments when reporting hasn’t made notice of a new requirement or if the information has reached the sponsor in a delayed fashion. Having automated processes in place prevents changes from falling through the cracks.
Subscribing to a regulatory database may be helpful but due to the issues occurring when countries do not issue a notice of changes, there still needs to be human oversight — a quality check of sorts — and interpretation into a practical ‘what needs to be reported, how and when.’ Again, this takes time and experienced resources, particularly when trials are being conducted in multiple locations and in multiple languages. While databases may be helpful, complexities still exist that require human intervention.
Making life easier
Automated reporting of SUSAR and other adverse events, based on each country’s regulations and which roles need to be notified within the CTMS clinical format preferred, can ease the regulatory burden.
While the responsibility of adhering to regulatory guidelines belongs to the clinical trial sponsor, the management of regulatory compliance documentation can vary from a sponsor’s in-house team to a CRO or other vendor that supports safety regulatory affairs.
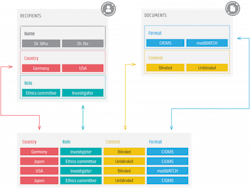
For example, a vendor specializing in pharmacovigilance can support the trial sponsor with an automated system, i.e., a dynamic data hub that is designed to manage contact information for recipients at all clinical trial sites, distribute safety documents uploaded or linked from existing data sources, provide audit trails as well as manage reporting requirements per country and per recipient type. In a typical safety document distribution scenario, ethics committee members would receive specific reports defined for each role and document type.
For the support of this distribution process, there are templates specifically designed per role and document type. These are based on local values and will apply to about 90% of all the rules but can be modified if necessary. Recipients are identified as to whether they should receive blinded or unblinded reports, which is critical when an unblinded report is inadvertently delivered to an unvetted recipient. This kind of mistake can seriously jeopardize a trial by creating bias. Data hubs can be updated when new contacts are onboarded or new safety documents are created — all of which are automatically entered into the system. The messages that are sent out are then tracked, with a detailed audit trail, allowing you to drill down from a document to see to whom it was sent and when it was acknowledged.
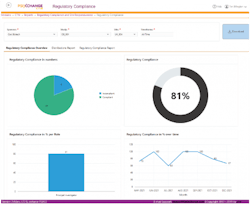
Utilizing the right compliance software platform can help ensure clear oversight of compliance status for all documents and manufacturing sites.
Most countries accept the standard report formats: MedWatch, CIOMS-I and CIOMS-II. The forms are generally distributed using English as a single language. However, specific processes or local forms are often translated into local languages, again which require translation and transparent interpretation. Another interpretation challenge may come from CROs operating in different countries where processes may be similar but not the same. Therefore, there is still human intervention involved for a quality check on accuracy and ‘clean-up’ of any errors.
Regulatory bodies in both the U.S. and Europe are offering directives to streamline reporting processes. In the EU, the regulations for drugs and medical devices have become far more aligned than they have been in the past. However, while regulations for safety document distribution often remain in place for several years, the reporting processes may change slightly, particularly in countries just learning to manage clinical trials, where laws may change more often.
The journey forward
It is within reach to implement completely automatic reporting options that vastly simplify the tasks involved in keeping track of regulatory process changes, which will positively affect the transparency in data reporting and greatly reduce labor, manual errors and their associated costs. The key to this journey forward is a basis in which the information flows are interactive and dynamic via a central dashboard, and where humans and machines work in tandem to assure prompt reporting with accurate results pre-determined to meet global guidelines.