Unpacking the Complexity of Biologic Drug Containment

The pace of biologic drug development, including the development of therapeutic proteins (especially antibodies) as well as gene and cell therapies, is accelerating. In 2017, 79 percent of approvals by the U.S. Food and Drug Administration of New Molecular Entities (NMEs) were biologic drugs and included both gene and cell therapy approvals. The complexity of these NMEs is increasing not only by size but also by function.
Beyond standard monoclonal antibodies (mAbs), more sophisticated approaches to drug design are imparting protein drugs with more intelligence and capability. As examples, antibodies conjugated to biopolymers are being investigated to extend the half-life of anti-VEGF drugs.1 Antibody-drug conjugates consisting of a small molecule covalently linked to the mAb, can selectively deliver cytotoxins to cancer cells.2 Antibodies may even be combined with nanoparticle delivery vehicles containing cytotoxic drugs so that the payload can be specifically delivered to mAb-targeted cancer cells. 3
While proteins, especially mAbs, are the most prevalent class of biologic drugs, gene and cell therapies have, in recent years, become a commercial reality and they represent a significant increase in therapeutic complexity. For example, a primary delivery challenge with gene therapies is that the nucleic acid drugs, designed to modify DNA to treat the disease, must pass through cell walls inside the patient’s body. Common strategies for transporting genetic material inside of cells include transduction by a viral vector or lipid-mediated transfection. This means the genetic modifier must be packaged into an adeno-virus or a liposome delivery vessel.
With the growing complexity of these therapeutic entities, it becomes more important to reduce product quality risks to the fullest extent during the entire life of the product from fill/finish to storage to shipping, and delivery to the patient.
While new biologic entities are evolving, the first-generation biologics are growing in ubiquity. As of June 2018, the FDA has approved 11 biosimilar products. In fact, five of the top 10 bestselling drugs in 2017 now have FDA-approved biosimilar counterparts including Humira, Enbrel, Remicade, Avastin and Herceptin.

Compounding Complexity
The fact that protein drugs are becoming so common means that they have firmly established their presence in the compounding world, bringing new challenges to 503A pharmacies and 503B outsourcing facilities who have traditionally handled mostly small molecules. One example is oncology infusion clinics which prepare and administer chemotherapy agents and which are compelled to operate in accordance with USP<800> guidelines for handling hazardous drugs. In 503A pharmacies that prepare patient-specific prescriptions for oncology infusion, pharmacists and healthcare workers routinely handle small molecule cytotoxic/genotoxic drugs in the same facilities by the same means as they handle biologic drugs.
While there is a small subset of biologics which may be cytotoxic, most are not considered to be toxic. Routes of accidental exposure present low risks for proteins — they are generally molecules that are too big to pass through the skin or be easily aerosolized, and they would be subject to enzymatic degradation in the case of oral exposure. Yet, despite the lower risk profile of recombinant protein therapeutics as compared to cytotoxic small molecules, the two therapeutic classes are stored and handled together in oncology infusion pharmacies.
These handling practices are designed to reduce the risk of exposure to hazardous drugs by maintaining one set of rules for all drugs. In the process, however, non-hazardous protein therapeutics could be exposed to a higher risk environment.
For 503B outsourcing facilities which handle non-patient-specific, larger-scale nationwide drug distribution, biologic drugs present a whole new set of challenges Compounders who have traditionally selected the most cost-effective containment options and who have not scrutinized drug stability or container interactions, are now being ushered into the more heavily scrutinized world of conventional parenteral packaging and the extensive characterization that comes therewith. This represents a substantial shift in the rapidly evolving compounding world and may leave some 503B outsourcing facilities struggling to build the required packaging expertise, relying on external consultants/test facilities, or even exiting the biologics repackaging business altogether.
Given this growing complexity, new challenges continue to emerge and drug manufacturers and compounders alike need to think differently about the packaging and delivery of these therapies.
Because these biologic entities are complex, their stability risks are higher and because they are large in size, their immunogenicity risk is higher when compared to small molecules. Therefore, the objective of manufacturers when it comes to packaging and delivery of biologics is to reduce risk to the fullest possible extent. Whether the challenge is to provide robust cryopreservation containment for gene or cell therapies, to develop wearable injectors to transition oncology mAbs out of infusion clinics and into a home setting, or to modernize the packaging systems used by 503B outsourcing facilities, there are several universal best practices for ensuring the quality of the biologic product.
Material Selection
Selecting appropriate materials for packaging biologics is the most basic consideration in reducing stability risks. Biologic drugs are complex entities which are susceptible to
instabilities when exposed to light, temperature changes, leachables, reactive gases, formulation changes, and other stressors. For this reason, most biologic drug manufacturers select materials which present the lowest possible risk of interaction such as barrier coated elastomeric closures and low surface energy container materials, while simultaneously seeking to minimize or eliminate silicone oil in these systems.
Especially in the compounding industry, the use of containers that are not intended for long-term contact (such as heavily siliconized polypropylene syringes) presents the risk of unforeseen stability issues4 and potentially shorter Beyond Use Dates (BUDs). The FDA’s guidance, states that each Biologics License Application (BLA) should include an analysis of the suitability of the container systems, the stability (of the drug in the container system and of the container system itself) and a quality control strategy.
While less stringent than the requirements for a BLA, the requirements for compounders (operating outside the scope of a BLA), are beginning to align more closely. In January 2018, the FDA issued a final guidance on “Mixing, Diluting, or Repackaging Biological Products Outside the Scope of an Approved Biologics License Application.” A noteworthy part of this guidance is Appendix A which, provides formal guidance for stability testing that is reminiscent of the expectations of a BLA. As examples, subvisible particle levels must be tested and USP<788> and USP<789> requirements are referenced. USP <1207> is referenced for establishing Container Closure Integrity (CCI) and the maintenance of sterility.
Also, product potency and impurities (including protein aggregates, charge variants, etc.) must be characterized in order to assign a BUD. Compounders may reduce stability risks and possibly extend BUD’s by moving away from low quality disposable packaging in favor of traditional parenteral packaging options.
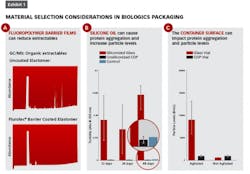
Exhibit 1 (left) shows several examples of impacts that material selection can have on attributes such as extractables and protein aggregation. In part A, gas chromatograms of an n-heptane extraction solvent that has come into contact with an uncoated elastomeric closure (top) versus a fluoropolymer film coated elastomer (bottom) show organic extractables originating from the rubber which can be blocked by the presence of the barrier film.
Some biologics, such as Orencia, as seen in Exhibit 1B, are prone to aggregation in the presence of silicone oil (the red bars show Orencia aggregation in siliconized glass syringes). By eliminating silicone oil altogether, the aggregation can be mitigated — as shown by the blue bars indicating unsiliconized cyclic olefin polymer (COP) syringes.
Finally, there is growing appreciation that the surface energy of the container can impact protein aggregation. In Exhibit 1C, vials made of glass (which has a highly negatively charged surface) are associated with higher degrees of aggregation of the selected model protein (with agitation and in the absence of surfactant) as compared to COP vials (which have a low surface energy). Selecting primary packaging materials with low surface energy (such as COP) and those materials which eliminate silicone oil may help drug manufacturers and compounders alike to meet USP<788> and USP<789> particle level requirements.
System Integration
Another best practice to ensure quality and reduce risks in biologic drug development is to consider, at the earliest possible stages, the packaging and delivery from a full integrated systems perspective. From the simplest container systems such as vials to the most complex delivery systems such as wearable injectors, all pieces of the packaging/delivery device should be designed and should operate as an integral system: The components must work in concert with the drug and with each other, under applicable storage and delivery conditions.
Most gene and cell therapies will initially go to market packaged in vials which are cryogenically stored at either –80ºC or –180ºC until delivery. Under these harsh storage conditions, polymer materials such as halobutyl elastomers used in vial stoppers will experience volume shrinkage in accordance with the material’s coefficient of thermal expansion (CTE).
The volume shrinkage is similar for rubber stoppers and for cyclic olefin polymer vials (approximately 4.5 percent and 4.1 percent respectively in transition from room temperature to –180ºC.) Glass however shows little volume shrinkage. The mismatch in CTE’s for rubber and glass presents a higher risk for container closure integrity breaches during cooling, storage, and warming as compared to the combination of rubber and COP. This has previously been observed experimentally5 through oxygen headspace analysis. Even in the simplest case of vial containment, it is critical to understand how the components of a packaging system work together and it is helpful if a packaging supplier can provide data packages to evidence the performance of the integrated system.
A drug manufacturer filing a combination product that uses an autoinjector to deliver a viscous drug must understand how the drug viscosity impacts the syringe delivery forces and therefore the autoinjector design requirements. If such data can be provided by the syringe manufacturer, it can help to de-risk the combination product development, reduce the time to market, and help to ensure the most reliable device performance.
Wearable injectors can deliver large drug volumes over an extended period of time and may offer a solution for moving oncology biologics out of the clinic and into a homecare setting. Reliable performance of such a complex device, is the culmination of successful system integration of the drug, primary packaging and delivery device.
With the growing complexity of new therapeutic biologic entities, it is becoming more important than ever for drug manufacturers and compounders to reduce risks. Choosing the appropriate packaging materials and considering full system integration at the earliest stages are two foundational best practices for ensuring quality. While accountability for the safety, compatibility and performance of a combination product lies solely with the drug manufacturer who is filing the drug with the regulators, or with the compounder who is repackaging the drug, responsibility for the full system characterization is becoming more broadly distributed across the supply chain.
References
1. V. Perlroth. Antibodies and Conjugates Thereof. US 2017/0190766A1, July 6, 2017.
2. A. Beck et al. Strategies and challenges for the next generation of antibody–drug conjugates. Nature Reviews Drug Discovery, 16, 315–337 (2017).
3. S.K. Singh et al. Drug delivery approaches for breast cancer, Int J Nanomedicine, 12, 6205–6218, 2017.
4. www.fda.gov/Drugs/DrugSafety/ucm458952.htm.
5. W. Winters. When Glass Vials Fail at Low Temperatures, Consider A Cyclic Olefin Polymer System.
Acknowledgements
Many thanks to Cathy Zhao, Lloyd Waxman, Ranjana Singh, Le Ho, Page McAndrew, and the entire West Analytical Laboratory Services team for their contributions to this work.
[javascriptSnippet]