Annual Product Reviews: How to Conduct an Effective Annual Product Quality Review

The GMPs necessitate annual evaluation of quality standards of a drug product to determine the need for adjustments in drug product specifications, manufacturing and control procedures. Subpart J of 21 CFR 211.180 mandates establishing a written procedure for the Annual Product Review process, while, and recommends review of a representative number of approved as well as rejected batches.
The guidelines stress the importance of analyzing investigations/deviations conducted and product complaints received, while the APR report must explore, in-depth, causes of recalls and returns, if any. Overall, FDA’s mandate is to look thoroughly and systematically for areas of improvement and to align processes to consistently manufacture quality products.
For these same general reasons, Canadian GMP’s were updated in 2009 to include an explicit section for Annual Product Quality Review (C.02.011). Health Canada’s regulations require manufacturers to analyze previous reviews, examine finished product testing results and critical in-process controls, and review: failed batches, deviations, CAPA effectiveness, changes, stability studies, returns, complaints, recalls, critical equipment qualifications, and quality agreements. CAPA’s from annual product reviews need to be communicated to senior management and completed in a timely and effective manner, with effectiveness verified via self-inspections.
And in the EU, Product Quality Review requires a review of starting materials including packaging materials used, a review of marketing authorization variations submitted/granted/refused, and a review of post-marketing commitments. The Qualified Person is responsible for the accuracy and timely completion of the review. Finally, the PIC/S GMP guide is in line with the above requirements.
Despite the similarity of these expectations, there are a few unique expectations, as shown in Table 1.
Table 1. APR Requirements Unique to Regulatory Authorities
| FDA | Health Canada | EU |
|
|
|
Structure of an APR Report
The structure of a review report can vary based on different products and a firm’s specific documentation requirements. Yet, manufacturers should follow a standard template to ensure that all required aspects are evaluated.
But an APR is also an evolving document. It can be of few sections with minimal requirements to an elaborate document with addenda containing information or data relevant to the product (e.g., retain sample review). Each numbered sub-section (batch document review, change review, deviation review, etc.) is typically followed by a summary. A graphical or tabular representation will help in dissecting data and detecting adverse trends wherever applicable.
A sample list of contents is contained in Figure 1, while Figure 2 shows a typical list of data tables.
| Figure 1 |
| 1. Scope 2. Manufactured Batches, Batch Record, Yield Review 3. Change Control Review 4. Label and Artwork Change Review 5. Analytical Data Review 6. Stability Data Review 7. Validation and Qualification Review 8. Non-Conformances and LI Review 9. Rejected Batches Review 10. Re-Packaged Batches Review 11. Compliant Review 12. Field Alert and Recall Review 13. Retain Sample Review 14. GMP Agreement 15. Review of Previous APR 16. Conclusions and Recommendations 17. References 18. Approval |
| Figure 2. Common Tables and Charts |
| • Summary of Lot Enumeration for Bulk Batches • Summary of Lot Enumeration for Finished Product • Summary of Batch Yield • Summary of Analytical Results • Summary of In-Process Results—Encapsulation • Reserve Sample Annual Inspection • Summary of Changes • Description of Deviations and Non-Conformances • Batches introduced to the Stability Program During Review Period • On-going Stability Batches introduced Prior to Review Period |
The scope of the review needs to explain the purpose and the product sku’s covered. Information from the batch processing and packaging records can follow. This includes review of in-process, process SPC charts, yields, analytical results, and so on, as applicable.
Changes and Corrections
Change review can be broken down to raw material changes, packaging component changes, master document changes and specification changes. The non-conformances/deviations section needs to review non-conformances but also corrective actions and their effectiveness. Any ineffective or overdue CAPA needs to be discussed in the summary.
The crux of the APR document is the Conclusions and Corrective Actions/Recommendations section. This section should include summaries of each of the prior sections, and the appropriate corrective/preventive measures necessary for each observation made. Any trend observed needs to be addressed, not just those that are out of specification or borderline. Figures 3, 4, and 5 are representative charts.
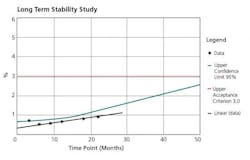
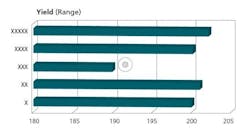

Streamlining Data Sources and APR Administration
Streamlining the entire process requires an APR schedule, based upon key regulatory submission dates. (For contract manufactured products, it’s critical to prioritize and negotiate feasible reporting dates.) Compiling APR raw data is always a team effort, but the Compliance/QA department should take the lead and be ultimately responsible for the program and its administration. An APR committee would typically include a representative from QA, QC, Validation, Operations, Stability, Engineering, and Materials Management. A draft report is completed upon critical analysis of the raw data, then discussed in APR committee meetings to determine effective CAPA’s.
Another challenge for the APR administrator is data retrieval for review purposes. Firms with qualified data acquisition systems can use their database, whereas paper-based manufacturers may have to review individual batch documents for processing parameters, in-process testing, finished testing, yields, and so on. In either case, the raw data used for analyses must be accurate in order to complete an effective assessment. If process drifts are observed during review, additional information may need to be collected to substantiate the findings.
Data must be available to the APR administrator for his/her in a timely fashion. They all must then be verified by a second person if performed manually. If spreadsheets are used, they must be qualified in advance.
Footnote
Performing an APR is a requirement for the regulated market. But more than this, the review helps the manufacturer to understand processes better and to gather additional information for further improvements. It greatly helps in determining if a product still meets the needs of patients, if it needs a formulation change, packaging modification, a revised specification, or a more robust process. An APR conclusion is stepping stone towards the future development of the product and hence should be accurate and backed by adequate data.
FDA’s Process Validation guidelines call for continued process verification. Thus, an APR program can serve as an ongoing system (Stage 3: continued process verification) to collect and analyze product and process data that relate to product quality. The APR needs to be part of the risk management/mitigation plan developed, per ICH Q9 recommendations.
The information gathered and trends spotted can aid new product development as well, and so it is essential to distribute the report to all relevant and interested parties. The effort can also be reviewed and shared with Lean process improvement teams, while the CAPA’s developed out of an APR are critical in avoiding potential risks to a product in the future.
References
1. 21 CFR PART 211, Subpart J, Sec. 211.180.
http://www.accessdata.fda.gov/scripts/cdrh/cfdocs/cfcfr/CFRSearch.cfm?fr=211.180
2. Health Canada Good Manufacturing Practices (GMP) Guidelines
- 2009 Edition (GUI-0001): Section C.02.011
http://www.hc-sc.gc.ca/dhp-mps/compli-conform/gmp-bpf/docs/gui-0001-eng.php
3. EU Guidelines to Good Manufacturing Practice: Chapter 1. Quality Management:
http://ec.europa.eu/health/documents/eudralex/vol-4/index_en.htm
4. FDA Process Validation: General Principles and Practices
http://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/UCM070336.pdf
5. ICH Q9:
http://www.ich.org/fileadmin/Public_Web_Site/ICH_Products/Guidelines/Quality/Q9/Step4/Q9_Guideline.pdf
6. ICH Q10:
http://www.ich.org/fileadmin/Public_Web_Site/ICH_Products/Guidelines/Quality/Q10/Step4/Q10_Guideline.pdf