Introducing PAT, Using NIR Analysis, to a Pharmaceutical Blending Process

This study investigates the use of a laboratory-scale blender fitted with a near-infrared (NIR) probe to monitor lubricant uniformity in a granule blend. A software method was developed to monitor the change in absorbance at significant wavelengths for the granule and lubricant (magnesium stearate) as the blend proceeded in real time. The standard deviation of the absorbance was plotted as a function of time to monitor the change in the blend. With near-infrared spectra, when a process is complete, the spectra will not change; therefore the standard deviation will be small6.
INTRODUCTION
Process analytical technologies are systems for the analysis and control of manufacturing processes to assure acceptable end-product quality11. This is achieved by timely measurements of critical parameters and performance attributes of raw material and in-process material and processes11. The desired goal of process analytical technology (PAT) is to design and develop processes that can consistently ensure a predefined quality at the end of the manufacturing process1. To build quality into a product requires the manufacturing process to be monitored and controlled as opposed to only testing the product at the end of the manufacturing process to assure quality1.
The blending of solids is a critical step in the production of many pharmaceutical products4, 7. Traditional methods for blend uniformity determination involve sampling of the blend using a sample thief and laboratory analysis of the samples using chemical methods2. In addition, blend uniformity in the traditional sense focuses on the distribution of the active pharmaceutical ingredient in the blend as opposed to distribution of the excipients in the blend, which can influence the desired performance of the pharmaceutical product2.
Magnesium stearate is a commonly used tablet lubricant that forms a film of low-shear strength around the granule, thereby reducing the friction at the die wall during tablet ejection8. Blending for longer durations than is necessary could result in the incorporation of magnesium stearate intra-granularly, which can influence the bioavailability by decreasing the dissolution rate of the product, due to the hydrophobic nature of magnesium stearate9. In addition, overmixing could lead to the physical destruction of the granule leading to poor compression profiles9.
The common problems associated with poor lubrication are: binding where tablets have vertically scratched edges, lack smoothness or gloss and are often fractured at the top edges; sticking where tablet faces appear dull; filming, which is the early stage of sticking; picking, which represents the advanced stages of sticking and capping; and lamination, which is normally associated with poor bonding and can also be a result of a system that is over-lubricated5.
Due to the competitive nature of the pharmaceutical industry and the continuous emphasis on quality from the regulatory authorities, pharmaceutical manufacturers require a system to monitor all materials in a blend with little or no sample preparation and the ability to build quality into the product and predict end-points in real time. PAT offers these advantages and near-infrared (NIR) spectroscopy is one such tool that is commonly used3.
The majority of active pharmaceutical ingredients and excipients absorb NIR radiation; therefore, NIR has the ability to provide information of all the components in the blend and is non-invasive, speedy and requires no sample preparation2. In this study, NIR spectroscopy was used for the on-line monitoring of magnesium stearate in a granule blend.
EXPERIMENTAL
Materials
Magnesium stearate (EP 5.02) from approved suppliers to the pharmaceutical manufacturer was used as the lubricant. A proprietary granule was chosen to mix with the magnesium stearate.
Equipment
The study utilized a laboratory-scale blender fitted with an NIR probe, operated by equipment specific software programs. During the intermediate bulk container (IBC) inversion, the NIR probe measured the NIR spectrum of the material in the IBC. Therefore, each and every rotation captured an NIR spectrum.
Obtaining fingerprint spectrum
The pure spectrum was obtained by placing approximately 50 g of each material on the Corona head and measuring the NIR spectrum. The NIR spectra of four random samples of magnesium stearate and six random samples of the granule were measured and the mean spectrum of each material was calculated to obtain a fingerprint spectrum. The fingerprint spectrum of magnesium stearate and the granule were superimposed.
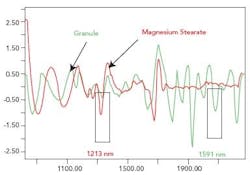
The significant wavelengths for blend monitoring for the materials were obtained by examining the spectra in the second derivative. The software was used to highlight the peaks on the spectra (as seen in Figure 1). Preliminary blends were conducted using the wavelengths obtained from the software. The criterion used to choose the most suitable wavelengths was that the standard deviation of the absorbance at the wavelength representing the magnesium stearate and the granule should increase when the magnesium stearate was added to the blend, and level off after a period of time. Following the preliminary blends using the wavelengths isolated by the software, 1213 nm was chosen to represent magnesium stearate and 1591 nm was chosen to represent the granule, as highlighted in Figure 1.
Next, a method was created on the software to interpret the spectral data in terms of the rate of change in the blend.
The second derivative was used for spectral pre-processing and the results were evaluated at the wavelengths of significance. The software was set to evaluate the standard deviation of the absorbance at the wavelengths of significance in a moving block of eight as outlined below.
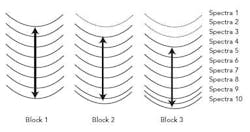
Figure 2 shows a schematic of the variance calculation. Spectra are added to the block until eight spectra are obtained (seen in block 1). Then, for every spectrum that moves out of the block, one spectrum is added to the block (seen in block 2 and block 3). As the spectral difference decreases, so too does the moving block difference. The standard deviation is calculated over the block and plotted over time to monitor the rate of change in the blend. With NIR spectra, once a process is complete, the spectra will not change, and therefore the standard deviation will be small6.
Spectral data from blends
During blending, spectral data were collected by the NIR probe during each rotation of the IBC. The IBC was set to move at 10 rotations per minute (rpm). The data were mathematically treated with the algorithms available on the software created during the method development. The blend was monitored by the standard-deviation-versus-time plot that was generated. To confirm the prediction of end-point using NIR, the blends were sampled using a sample thief when the NIR spectrum showed no more change with respect to time at the wavelengths of significance (when a plateau level was reached) and at selected time intervals.
In order to determine the time intervals at which to sample, preliminary blends were conducted. A series of blends were run for approximately 20 minutes each. The standard deviation was plotted as a function of time and examined to identify three time intervals in the blend to predict three states, namely before end-point, end-point and after end-point. The first time interval chosen was when the standard deviation had not leveled out and was high (depicting before end-point). The second time interval chosen was when the standard deviation was lower than the first time interval (depicting end-point) and the third time interval chosen was when the standard deviation maintained a low value for a longer period of time (depicting after end-point). Following these preliminary experiments, the times for sampling were obtained.
The intervals for sampling of the granule blend were one minute and 30 seconds (before end-point), six minutes (at end-point) and 17 minutes (after end-point). The blends were carried out in triplicate at the predetermined time intervals. Five point samples were taken from the blender according to a sampling plan and were analyzed using an atomic absorption (AA) spectroscopic method for magnesium stearate. One sample per point was taken due to the decreased capacity of the blender.
The standard deviation that predicted a uniform blend was obtained from the blends conducted to the selected time intervals. Six additional blends were run and stopped when the standard deviation was reached.
Blend preparation
Granule (3 kg) was loaded into the IBC. The IBC was rotated for eight rotations. Thereafter, the blender was stopped and 22.18 g of magnesium stearate was added to the granule. Care was taken to ensure that the magnesium stearate was evenly spread over the top of the granule. The IBC was then closed and blended for the predetermined time intervals. The blend was then sampled using a sample thief and the stated sampling plan.
Limits for AA results
A unit dose sample is 102.2 mg, of which 0.75 mg is magnesium stearate. The United States Pharmacopoeial (USP) limit of content uniformity was used to calculate the acceptable limits for magnesium stearate10. The criteria for a uniform blend was a magnesium stearate assay of 0.6375 mg 0.8625 mg and a relative standard deviation (RSD) less than or equal to 6.0%.
RESULTS AND DISCUSSION
As the wavelength of significance for magnesium stearate was shown to be 1213 nm, the change in absorbance at this wavelength is presented. The standard deviation versus the number of rotations at the magnesium stearate wavelength for blends conducted for one minute and 30 seconds depicting before end-point can be seen in Figure 3.
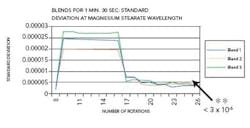
An increase in the standard deviation was noted when magnesium stearate was added to the blend. At the end of the blend duration, the standard deviation was above 3 x 10-6. The AA results revealed a non-uniform distribution of magnesium stearate.
Figure 4 reflects the blends carried out for six minutes, depicting end-point.
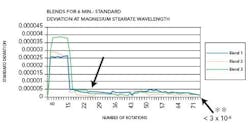
A leveling out of the standard deviation could be seen at the 28th rotation. The standard deviation at the magnesium stearate wavelength was less than 3 x 10-6 at the end of the blend duration and the AA results revealed uniform distribution of magnesium stearate in the blend. Figure 5 represents the blends conducted for 17 minutes depicting after end-point.

A leveling out of the standard deviation was noted at the 28th rotation. At the end of the blend duration, the standard deviation at the magnesium stearate wavelength was less than 3 x 10-6. The AA results revealed uniform distribution of magnesium stearate in the blend. Table 1 shows a summary of the relationship between the standard deviation and the uniformity of the blend for blends conducted to predetermined time intervals.
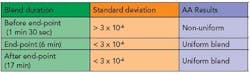
The distribution of the magnesium stearate in the blends for 17 minutes and 6 minutes show a relationship, in that both sets of blends are uniform and the standard deviation of the absorbance was below 3 x 10-6. It was noted that a decrease in blend time of 11 minutes still resulted in a uniform blend. The blends for 1 minute and 30 seconds had a higher standard deviation (greater than 3 x 10-6) and the AA results revealed an uneven distribution of magnesium stearate.
Based on the blends conducted above, the criteria selected to predict a uniform blend in real time was a standard deviation value below 3 x 10-6 at 1213 nm (wavelength for magnesium stearate) at four consecutive data points. Figure 6 shows the standard deviation versus number of rotations for six blends conducted to an end-point as predicted by NIR and Table 2 shows the AA results.

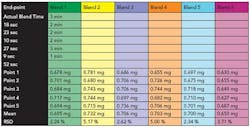
The results show that all blends were uniform with the exception of blend 6 where the amount of magnesium stearate was marginally out of specification. Blend 6 also represents the shortest blend duration. It was noted that the standard deviation at the 1591 nm (wavelength for granule monitoring) wavelength at the end of the blend was the highest for blend6.
At present, the blend duration for this granule is set at 10 minutes, which was determined by standard validation techniques. With NIR, blend durations range from 2 minutes 10 seconds to 3 minutes 18 seconds. It was noted that the time durations are significantly lower than the standard blend durations.
CONCLUSION
A relationship exists between the uniformity of the blend and the standard deviation of the absorbance at the significant wavelength. Blends were found to be poorly blended when the standard deviation of the absorbance was high and well blended when the standard deviation of the absorbance was low.
It is evident that more information regarding the uniformity of the blend could be obtained from the 1591 nm wavelength to aid an accurate prediction of end-point using NIR. The standard deviation value selected should be on a product-by-product basis and optimized in conjunction with standard blend-uniformity methods.
ACKNOWLEDGEMENT
The authors would like to thank Aspen Pharmacare for the supply of all materials and the sponsorship of equipment utilized for the study.
About the Authors
Krishel Naicker and Gareth Kilian (e-mail: [email protected]) are with Nelson Mandela Metropolitan University, Department of Pharmacy, Faculty of Health Sciences, PO Box 77000, Port Elizabeth, 6031, South Africa. Johan Olivier is with Aspen Pharmacare, 7 Fairclough Road, Korsten, Port Elizabeth, 6014, South Africa.
REFERENCES
1. Afnan, A.M. 2004. PAT: Whats in a Name? The Journal of Process Analytical Technology [online], 1(1), pp. 8-9. Available: www.patjournal.com/article.aspx?article=8 .
2. Ciurczak, E. W. and Drennen III, J. K. 2002a. Ch. 3: Blend uniformity Analysis, in Pharmaceutical and medical applications of near infrared spectroscopy, vol. 31, E. W. Ciurczak and J. K. Drennen III, eds., Marcel Dekker Inc, New York, pp. 33-54.
3. Ciurczak, E. W. 2006. Near-infrared spectroscopy: Why is it still the number one technique in PAT? The Journal of Process Analytical Technology, vol. 3, no. 1, pp. 19-22.
4. El-Hagrasy, A. S., Morris, H. R., Damico, F., Lodder, R. A. and Drennen III, J. K. 2001. Near-infrared spectroscopy and imaging for the monitoring of powder blend homogeneity. Journal of Pharmaceutical Sciences, vol. 90, no. 9, pp. 1298-1307.
5. Gilbert, S. B. and Anderson, N. R. 1986. Tablets, in The theory and practice of industrial pharmacy, 3rd edn., L. Lachman, H. A. Liberman and H. L. Kanig, eds., Lea and Febiger, Philadelphia.
6. Hammond, S. 2005. PAT strategies for identifying, measuring and controlling process risk: Process to measurement interface issues- Set up and remote triggering [online]. Available: www.fda.com.
7. Lantz, J. and Schwartz J.B. 1989. Ch. 1: Mixing, in Pharmaceutical Dosage Forms: Tablet, 2nd edn., H.A. Liberman and J.B. Schwartz, eds., New York: Marcel Dekker Inc, pp 1-71.
8. Marshall, K. 1986. Compression and consolidation of powdered solids, in The theory and practice of industrial pharmacy, 3rd edn., L. Lachman, H. A. Liberman and H. L. Kanig, eds., Lea & Febiger, Philadelphia, pp. 66-99.
9. Muzzio, F. J., Alexander, A., Goodridge, C., Shen, E. and Shinbrot, T. 2003. Solids mixing, in Handbook of industrial mixing: Science and practice, E. L. Paul, V. Atiemo- Obeng and S. M. Kresta, eds., Wiley & Sons Inc., New York, pp. 906-907.
10. Pharmacopeial Forum. 2006. Ch. 905: Uniformity of dosage forms, in USP29-NF24. USPC, Inc. Official.
11. United States Food and Drug Administration, United States Department of Health and Human Services, Centre for Drug Evaluation and Research, Centre for Veterinary Medicine and Office of Regulatory Affairs. 2004. Guidance for Industry: PAT: A Framework for Innovative Pharmaceutical Development, Manufacturing, and Quality Assurance. Available: http://www.fda.gov/cder/guidance/index.htm.