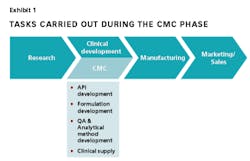
Evolution in the Era of New Biopharmaceuticals
1) Contact with regulatory agencies
Why is it important? While continuing dialogue with regulatory agencies is needed for all molecular formats, the relative novelty of both biosimilars and complex drug product formulations mandates an even closer relationship than normal. The uniqueness of these formulations implies that the regulatory agencies are likely not to know at the outset exactly what kind of scientific proof is needed for a product to be “good enough” to ensure patient safety. Companies should expect agencies to be exceedingly cautious in granting approvals, making it imperative that the CMC unit stay informed throughout the development process about what evidence will be needed so that it won’t be blindsided by unexpected requirements.
The original guidelines for biosimilar development regulation came from the European Medicines Agency in 2005 and the abbreviated pathway for biosimilars included in the U.S. Affordable Care Act in 2010. The rules, however, are still new, and more biosimilars need to enter the market before better clarity will be established regarding the requirements to prove biosimilarity.
Drug delivery systems pose a different concern for health authorities because many of these systems add an extra layer of complexity regarding product formulation that is difficult to characterize fully. So that no time is wasted in CMC, frequent communication with health authorities will be needed early in the process to ensure that the analytical development satisfies regulatory needs.
A key point, emphasized by the FDA, is that the evidence needed for approval of either biosimilars or complex drug delivery formulations may differ on the basis of the pharmaceutical drug. There will be a core set of analytical data expected in the CMC filing and additional analysis on a case-by-case basis that differ significantly among assets. In proving biosimilarity, safety or efficacy, however, the sufficiency of the additional analyses needed is assessed on a case-by-case basis by the specific regulator. A good example is the evidence burden needed to prove biosimilarity between a relatively simple insulin molecule consisting of 51 amino acids not glycosylated versus proving biosimilarity between a mAbs consisting of approximately 1,300 amino acids with multiple glycosylations. The latter will clearly need more evidence to prove similarity. Ensuring that the experimental setup is robust enough through early and frequent dialogue with the health authorities can potentially reduce the need for further clinical trials.
How to do it? Early dialogue with the regulator, specifically on CMC of an asset, helps ensure that the right input is available in a timely fashion. It is important that the regulatory representative responsible is an expert in CMC and quality issues, and not just in clinical development. Regulatory inputs need to be worked into the CMC strategy for the product, necessitating ongoing interaction between the regulator and the CMC representative. This can be best facilitated through a CMC project team.
2) Quality by Design
Why is it important? Biopharma manufacturers have conventionally defined a manufacturing process and sought to perform that process consistently in order to ensure that critical parameters are kept within a narrow range of specifications. This approach has produced safe and efficacious biotechnology products. Any variability, however, in raw materials, environmental controls and so on will result in variability of the product that can potentially lead to batch failure. QbD is an approach to control these problems. Initially advocated by the FDA in its process analytical technology guidance, QbD is intended to yield a better understanding of how variability in process parameters ultimately affects product quality. Ultimately, QbD provides insights into the process parameters that matter most. Having this knowledge builds comfort that the process and the product quality can be effectively controlled. QbD has implementation costs, but its long-term business potential in terms of cost savings and faster time-to-market is very significant (T. Fuhr, 2010).
For complex drug delivery formulations and biosimilars, the advantages of QbD become even more pronounced.
The parameter space for a complex drug delivery formulation is much more complex than for a simpler biopharma product. Liposomes or other nanoparticles, for example, have many process parameters determining shape, size and distribution of the particles, such as formulation temperature profile and stirring speed. Given the vast number of process parameters, it is critical to understand which process parameters have the greatest effect on product quality so as to ensure low variability of the finished product’s characteristics. There’s another big benefit to QbD: having the parameter space analyzed for quality effect on the product will ease the process of persuading the FDA to approve the product even though only a few previous cases are available for comparison.
For biosimilars, QbD will be critical in ensuring that production costs are low and that time to market is fast by avoiding quality issues that appear in the scale-up process. Having a set of well-known cell lines that serve as “platforms” for biosimilar development could prove to be a viable strategy. With platforms, knowledge of critical parameters affecting product variability and yield can be built in upfront, independent of the molecule being developed, thus easing the task of creating needed QbD data.
In both cases, the arguments for implementing QbD are strong with respect to quickly gaining in-depth knowledge and control over of the drug substance and drug product development.
How to do it? It is important to have the right mindset when implementing QbD. It should be seen not as an extra burden mandated by increasing regulatory requirements but rather as an opportunity to learn, ensure smooth scale-up and simplify future manufacturing operations.
The first step in effectively implementing QbD in biopharmaceuticals is to ensure that the expertise is available and being continuously developed within the organization. To ensure such expertise, a CMC organization needs the right talent and to proactively manage knowledge sharing and training. Secondly, it is necessary to develop a tailored QbD approach to each asset. Completely standardized QbD approaches are likely to be too expensive and not acceptable for all assets, although introduction of tech platforms and standardization will significantly simplify efforts.
3) More analytical capabilitiesWhy are they important? Relative to originator drugs, biosimilars will typically be produced in different cell lines or under different environmental conditions. They thus may be expressed with subtle but important differences. The FDA believes that three protein properties are highly relevant to these potential differences and must be analyzed: post-translational modifications, three-dimensional structure and protein aggregation. The challenge, however, is that they cannot be measured effectively enough to provide proof of similarity beyond legitimate doubts. Hence, the quest for a continuous analytical method improvement continues.Using liquid chromatography-mass spectrometry (LC-MS) methods, the primary sequence and the presence and identity of post-translational modifications can be determined. Characterization of the glycosylation present on recombinant proteins is particularly important because the carbohydrates attached to the molecule can modulate the stability and ability of the molecule to elicit its desired effector response.But slight, nondetectable differences in molecular composition may lead to significant changes in higher-order structure. In fact, many of the forces holding the structure together are weak, noncovalent interactions. To analyze higher-order structure, similarity methods such as circular dichroism, fluorescence, differential scanning calorimetry, analytical ultracentrifugation, and size exclusion chromatography can be used. These analyses, however, do not provide information on which locus on the molecule may give rise to a difference between biosimilar and originator molecules. Protein structural analysis using advanced experiments such as hydrogen-deuterium exchange MS has begun to yield greater insight as to where on the molecule changes do exist and hence to make a real case regarding similarity.A different set of analytical capabilities are needed for more complex drug delivery formulation because the formulations typically will comprise a distribution of particle sizes and composition. As a result, the focus is on proving a narrow distribution of the formulation and reproducibility from batch to batch versus proving a unique identity with a high purity. To investigate the size distribution, ensemble techniques such as dynamic light scattering and circular dichroism and single-molecule techniques, such as electron microscopy, are powerful tools to master in order to show that the distribution is narrow and consistent.How to do it? As regulator expectations will progressively increase; companies will be expected to constantly have products and processes that are characterized by the best possible levels that gold-standard analytical technology will allow.The imperative is to remain at the forefront of the analytical technology evolution. The implication is the need for an explicit strategy on how to nurture internal, and access external, capabilities. Comprehensive levels of intelligence about which analytical methods exist and are being developed must be mapped against the product pipeline. Given the number of pipeline assets demanding a certain analytical capability, a decision should be made regarding internal development or outsourcing. A contract manufacturing organization is a good choice for noncore technologies. In-house analytical capabilities are the right choice for the analytical technologies in which the CMC organization may have scope to build internal expertise given the pipeline size and composition.
4.) Seamless handoffs and the use of CROS/CMOs
Why are they important? In conventional biopharmaceuticals, there is a handoff of knowledge on the drug substance from the research unit to the CMC functions. This information is built up through a process of target screening, selection and cloning.
When novel drug delivery formulations make the drug product more complex, however, considerable drug product development must be performed in the research phase because formulation is a key driver of both efficacy and safety. In other words, drug substance and drug product will be codeveloped. This may be a challenge because capabilities are generally fragmented among different parts of the organization, with research leading the expertise on development of the biopharmaceutical’s active pharmaceutical ingredient and CMC holding the expertise on biopharmaceutical formulation. Conversely, in the case of biosimilars, the bulk of the formulation development will typically be done in the CMC unit, so the knowledge transfer for generating the drug product will not be that extensive.
Codevelopment of drug substance and drug product in the research phase adds a new dimension to knowledge transfer complexity because it is vital that information about the interplay between drug “substance” and “product” is flawlessly coordinated. High performers take the radical approach of organizing around projects so that they bring to bear all relevant competencies and functions; some, even more radically, create a merged organizational unit that specializes simultaneously in both drug substance development and drug product development.
At the other end of development, with the handoff to production, there must be an equivalently seamless knowledge transfer. The manufacturing organization needs to be involved early to ensure manufacturability of the product. There are added reasons for early involvement with novel formulations, like the imperative to know the effect of scale, which will have a large impact on choices that need to happen in early CMC. Similarly, because many of the technologies will be new, manufacturing needs to be educated by CMC on such technologies and the related processes.
Because many of the technologies needed for these novel formulations are not always available in-house, CROs/CMOs might be involved in developing some aspects of the final product. Taking some of the development outside of the company puts high demands on both parties in the collaboration. For example, the CRO/CMO will be required to carefully document the experimental evidence in development, so that when the molecule is handed back to manufacturing, the right knowledge will be in place to ensure manufacturability. The point is particularly critical when outside resources are used, because manufacturing cannot reach out to the development team as easily as with an in-house development.
For biosimilars, the handoff challenge is different. Because the drug is already known, the research organization does not perform many of the typical activities done for innovative drugs. The main tasks prior to CMC are determining the primary sequence and modification pattern of the originator molecule and cloning that sequence into a suitable host to produce the molecule. In cloning, it is preferable for the host to be one with which the company has previous experience, so as to allow faster analytical characterization and more rapid scale-up. In biosimilar R&D, the research part is limited and does not require the share of work related to product innovation. Hence, the full focus is on the CMC, making the case for pure biosimilar players to integrate into the CMC organization the few CMC capabilities that are generally anchored in the research part of the organization.
How to do it? An organizational decision must be made about whether the unique skill set needed to develop drug delivery formulations requires the drug product development team to follow the molecule into the CMC process. The handoff/no-handoff choice will be influenced by the number of these “unique skill set” molecules going through the pipeline. If the number is small, the drug product team should follow the molecule to ensure capability continuity in the technically difficult area of drug delivery formulation. If not, when there is a handoff, it will be more extensive than usual. This is because the knowledge of the drug product development is much more complex, demanding a differentiated approach compared with conventional handoffs. A formal handoff of responsibility linked to development milestones does exist, but it is coupled to the commitment of research, CMC, and manufacturing to remain substantially involved throughout the molecule’s development journey.
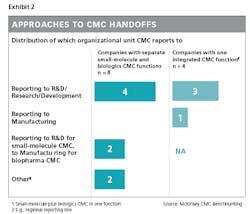
An example of how companies can strengthen handoffs to or from CMC is by having the CMC report in to either R&D or manufacturing, depending on where the most crucial handoffs are seen and where the CMC function can best leverage the existing skill set.
Setting up the CMC Function for Success
In summary, the new biopharmaceutical era presents challenges and opportunities to CMC units. To succeed, CMC organizations need to focus on three things:
• Improve capabilities throughout the function with respect to regulatory relationships, QbD and analytical technologies
• Ensure that tightly integrated teams are working in organizational setups that guarantee seamless handoffs between research and CMC, and between CMC and manufacturing or that are organized on the basis of product-focused cross-functional teams
•Integrate regulatory input and QbD early in the process of defining CMC strategies and plans
The question facing company leaders is, how can we accomplish these things in an efficient, effective manner?
The solution is to approach CMC like a project organization. The right mix of competencies is important, but even before that comes planning and adequate resource commitment. In this respect, CMC is no different from any engineering or product development project organization. But there is an additional complication: the risk of clinical development failure.
Given the unpredictability of clinical development success, CMC organizations are exposed to fluctuating workloads. Resource needs will be difficult to forecast, all the more so if the unit is pursuing several different molecular formats that require specialized technologies, capabilities and processes. The situation gets worse for those organizations whose pipelines do not have sufficient scale in the pursued molecular formats.
The mark of the effective, well-managed biopharma CMC unit will be its ability to flexibly optimize resources: to power up the right resources to swiftly develop biopharmaceuticals when the pipeline is full and to shed extraneous resources to avoid waste when it is not. CMC will be “living the trade-off” in a space where risk is coupled to critical fast-to-market requirements.
Success will depend on having the right organization setup and resources to finely balance those choices.
References
Ravetch, F. N. (2008). FC receptors as regulators of immune responses. Nat Rev Immunol, 8 (1) 34-47.
Philo, J. (2009). A critical review of methods for size characterization of non-particulate protein aggregates. Curr Pharm Biotechnology, 10; 359-372