10 tips for improved drug development

Bringing innovation to patients is no small task and a journey with many challenges.
Despite the disruption caused by the COVID-19 pandemic, the total number of products in active development globally exceeds 6,000, across a wide range of disease areas.
One measure of clinical development productivity involves relating molecule success rates to the complexity and duration of clinical trials. The composite success rate across all therapy areas — which describes the likelihood of bringing a drug candidate through regulatory approval — reached a ten-year low of 5% in 2021.
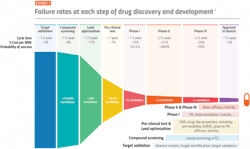
Efficiency of the R&D process must also be taken into consideration. The time from when a company first patents an innovation to new medicine launch represents a useful proxy for measuring R&D efficiency. The median time from first patent filing to launch for U.S. novel active substances was 10.7 years in2020 and 11.9 years in 2021, thus the time remaining to commercialize innovation until patent expiration decreased significantly.
Given these factors, the pharma industry should examine potential strategies to improve clinical development productivity and success rates.
1. Improve the drug optimization process
Analyses of clinical trial data from 2010 to 2017 show that 90% of clinical drug development efforts failed during phase 1-3 clinical studies due to four possible reasons: lack of clinical efficacy (40%-50%), unmanageable toxicity (30%), poor drug-like properties (10%-15%), and lack of commercial needs and poor strategic planning (10%).
This high failure rate raises the question of whether certain aspects of drug development are being overlooked.
But improving target validation and drug optimization — the process by which the best lead drug candidates are selected — has the potential to yield better results for the parameters mentioned.
One proposed solution to improve the drug optimization process is a structure tissue exposure/selectivity–activity relationship (STAR) system. This system classifies drug candidates into four different classes based on three aspects: 1) drug potency/specificity (high or low), 2) drug tissue exposure/selectivity (high or low) and 3) required dose for balancing clinical efficacy/toxicity (high or low). The four different classes of drug candidates (classes I-IV) require different strategies to select lead drug candidates, optimize clinical doses and balance clinical efficacy/toxicity.
The STAR system, therefore, takes an additional element of drug activity into consideration compared to the current conventional system.
2. Engage in earlier discussions with regulators
As a drug makes its way through the development life cycle, one of the most important relationships for a sponsor to maintain is with the Food and Drug Administration.
Expedited programs have been created for therapies that address the unmet medical need in the treatment of serious or life-threatening conditions. The choice of designation and timing of the application is critical to a development program.
These expedited programs also allow for informal, non-binding meetings with the FDA. These meetings are most effective when preliminary preclinical data — suchas proof-of-concept, and initial safety studies — have been collected. This allows the sponsors and regulators to proactively identify any scientific, legal or regulatory issues related to the chemistry, manufacturing and controls (CMC), pharmacology/toxicology, and/or clinical aspects of the development program. These meetings also help to prepare sponsors to derive maximum benefit from the available accelerated regulatory pathways.
3. Deploy novel trial designs
FDA guidance allows for the appropriate use of ‘adaptive designs’ for clinical trials to provide evidence of the effectiveness and safety of a drug or biologic. An adaptive design is defined as a clinical trial design that allows for prospectively planned modifications to one or more aspects of the design based on accumulating data from subjects in the trial. Adaptive designs can make drug development more efficient and less costly.
In addition to adaptive designs, the FDA has also initiated complex innovative trial designs (CID), which may provide potential benefits across a range of therapeutic areas. According to the FDA, there is no fixed definition of CID because what is considered innovative or novel can change over time, but CID includes trial designs that have rarely or never been used to date to provide substantial evidence of effectiveness in new drug applications or biologics license applications.
In both cases, some sponsors have made use of these available options with successful outcomes.
Former FDA commissioner Scott Gottlieb stated, “Using more modern approaches to clinical trials, we can lower the cost of developing new drugs.” But have these options helped to solve R&D productivity issues?

Mahlich et al. argued that it is reasonable to assume that a broader acceptance of adaptive trial design could improve R&D productivity by lowering attrition rates of phase 3 trials. They estimated that if attrition rates could be reduced from 38% to 20%, overall clinical success rates would leap from 11.8% to 15.8%. This would reduce the costs per new drug by 14%. In addition, they believe that their estimates are conservative because they did not consider efficiency gains that occur in earlier clinical phases.
4. Improve drug development through human genomics
It is an established fact that the majority of diseases have a molecular or genetic etiology. Some conditions, including sickle cell disease, cystic fibrosis, muscular dystrophy and Huntington disease, are caused by single-gene mutations. Syndromic conditions such as diabetes and cardiovascular diseases have multifactorial causes including multiple gene mutations confounded by environmental and lifestyle factors. In the context of drug discovery, genes have therefore been classified as disease genes, disease-modifying genes and druggable genes.
A genomic approach to preclinical target validation has the potential to reverse the current trend of low probability of drug development success when compared to the established (non-genomic) approach.
Recent trends in pharma R&D show that genomics may be employed in the recruitment of study participants for clinical trials with the selection favoring those subjects more likely to benefit from the intervention being trialed. This ensures that the effect of the drug will be evident if the drug is indeed effective against the target disease and absent if ineffective.
Genomics can also be used as a predictive tool to forecast potential toxicities emanating from a specific molecule, providing the opportunity to select trial participants who will best respond to the treatment being studied.
5. Utilize real-world data to inform trial design
Historically, real-world data (RWD) was used in the latter part of product development, often when phase 3 clinical trials were well underway. Increasingly, companies are leveraging RWD earlier in the development process in ways that are intimately tied to strategy. RWD and real-world insights (RWI) can be used to develop key portions of Target Product Profiles (TPPs), which help guide internal decisions throughout the product development process.
RWD can also be used by clinical pharmacology functions to inform the choice of medications for drug-drug interaction (DDI) studies based on the frequency of use in the patient population of interest. Information about comorbidities gleaned from RWD can also help assess the potential impact of DDIs based on renal and hepatic impairment and other health conditions.

RWD can also provide insights into potential subgroups of interest within the target indication byhelping predict response to therapy or identifying those with the greatest unmet needs according to the use of available therapies. Certain sources of RWD (i.e., claims data) can also inform estimates of the burden of illness (e.g., healthcare costs, impact on productivity, and mortality), which are key to developing market access, pricing and health economics and outcomes research (HEOR) strategies, such as value-based or outcomes-based contracts.
RWD help inform the design of more efficient clinical trials by better informing global target enrollment sizes needed, selecting more productive trial sites, enriching trial populations with predicted responders and increasing the diversity of trial participants.
6. Improve patient identification, recruitment, enrollment and retention
Patient enrollment and retention are a constant concern. Screen failure rates (determining who is eligible to enroll in studies) are significantly costly for sponsors and the average cost across the industry is roughly $1,200 per failure.
Approximately 80% of clinical trials fail to meet enrollment timelines, and around one-third of phase 3 clinical studies are terminated because of enrollment difficulties. Nine out of 10 trials require the original timeline to be doubled to meet enrollment goals.
Many variables impact patient visits and the quality of data reporting. Often these variables are burdensome on the patient and lead to increased patient dropout rates which, in turn, cause clinical study delays and inaccurate results. Most of the time, these issues are mitigated by identifying sites with higher patient retention performance scores in addition to understanding the patient enablement process implemented in a decentralized clinical trial approach.
For every 100 patients identified or available to move to the pre-screen step, only 7 patients complete the trial. On average, it costs $6,533 to recruit one patient for a clinical study, and the cost of replacing patients is even higher. The average cost to recruit a new patient if one is lost due to non-compliance is $19,533.10 These figures alone demonstrate theimportance of patient retention, but direct financial losses aren’t the only cost of patient dropouts.
7. Reduce the time between phase delays
On average, new drugs spend 40% of their development time in ‘white space’ — the time between clinical phases 1, 2 and 3.1 IQVIA notes four primary causes of between-phase inefficiencies:
- The time-consuming back and forth with regulators and health authorities
- Mistakes sponsors make in gathering inappropriate and incomplete inputs into decision-making late in the process
- The failure to anticipate downstream needs (e.g., manufacturing of clinical trial supplies)
- The inability to build the appropriate level of internal consensus and alignment around key decisions
- Addressing these inefficiencies early in the process can help reduce the white space.
8. Minimize protocol amendments
A study by the Center for the Study of Drug Development (CSDD) used data from 15 pharmaceutical companies and contract research organizations to establish price tags for the cost of implementing clinical trial amendments.
The study found that each amendment for a phase 2 trial cost a median of $141,000. For a phase 3 trial, the median cost was more than triple that at $535,000. The figures only include direct costs — for instance, additional fees to institutional review boards and increased costs to existing vendors for change orders — so the authors estimate the actual cost, including indirect costs, is likely three to four times larger than the direct cost for phase 3 trials, especially if the amendment delays the time to get to the market.
9. Capture data and analytics in clinical trials in real time
The adoption of today’s digital technologies offers a unique opportunity to revolutionize clinical trials with significant improvements in time, cost and the quality of data collected through the introduction of real-time data capture.
Pharma companies are investing in several technologies (e.g., electronic informed consent, and direct data entry into tablet computers), and remote patient monitoring (e.g., wearable or home-based medical devices transmitting patient data securely) that will enable real-time data capture and analytics.
10. Embrace and implement strategic new product planning
Dowden and Munro documented that poor strategic planning, which may include changes in therapeutic focus, company mergers or poor clinical studyconduct, accounts for 10% of drug development failures.
Success rates are crucial for R&D productivity because of the high cost of studies. Exceeding the industry-level success rate in each phase of clinical development will require a deep understanding of the drivers of the low success rate for each company and each development program; and a significant paradigm shift in the planning and execution of discovery and development.