Empty Chamber Studies (aka Much Ado About Nothing)
Empty chamber studies have been a part of sterilization validation since its beginnings in the 1970’s. The first regulatory mention of temperature distribution studies in the United States can be found in FDA’s draft Large Volume Parenterals (LVP) CGMP regulation from 1976 (never made official and withdrawn in 1991). Section 212.244 of the draft included the following requirement:
“The following factors shall be included in the design of sterilization processes: (a) Procedures required toestablish uniform heat distribution in the sterilization medium of the vessel. Such procedures shall be capable of holding temperatures throughout the sterilizer to within ± 0.5°C or ± 1 °F from the time the product achieves process temperature until the heating portion of the cycle ceases.”
The intent of this expectation was to minimize the temperature variation across the sterilizing chamber during the sterilization dwell period so that the exposed product containers are neither under- nor over-processed. In the execution of terminal sterilization validation studies this is still the expectation, however definitive numerical expectations for the empty chamber other than in the 1976 212 "regulation" have never been provided.
Industry’s response to the proposed regulation in the 1970’s brought into being practices that have shaped the execution of sterilization validation ever since. The first adaptation of the 212.244 "requirements" was to apply an identical metric to empty chamber studies. The logic in doing so was straightforward, if the chamber wasn’t sufficiently uniform without the load present; there was little value in proceeding to loaded chamber distribution studies where even greater variation could be anticipated.
While the 1976 regulation was intended for LVP’s, the Small Volume Parenteral (SVP) industry almost universally adopted the draft regulation for use with their moist heat sterilization processes, whether for terminal or in-process sterilization. The SVP industry accepted the "regulation" almost untouched, and many of the components of the 1976 document can still be found in present day sterilization validation.
Empty chamber studies typically precede load studies during initial qualification / validation and are frequently used as a part of change control evaluation. Over time, sterilizer performance has improved substantially, and even the largest terminal sterilizers routinely demonstrate substantially tighter temperature distribution ranges than were proposed some 40+ years ago. Adaptations have been made to incorporate empty chamber studies for sterilization-in-place, dry heat, gas and other sterilization process qualification/validation.
With such an auspicious beginning, it might be expected that empty chamber studies would be a definitive activity integral to sterilization qualification/validation. Surprisingly that is not the case. There are no formalized methods or acceptance criteria for the execution of empty chamber studies in conjunction with any sterilization process found in current regulatory or compendial standards! So despite its regulatory origins and prevalence within the healthcare industry, there is currently no "official' means to conduct an empty chamber study. This document endeavors to present some recommendations for their execution based upon my more than 40 years of experience with sterilization processes.
APPLICATION
Empty chamber studies are employed with steam sterilization equipment in at least three distinct ways:
1) As a User Requirements Specification (URS) element in the purchase of new sterilization equipment. This document establishes the performance expectations for new purchases. The restrictive steady-state temperature ranges desired in terminal sterilization come at a premium, so sterilizers for less critical activities, i.e., sterilization of laboratory media can utilize less exacting criteria.
2) As first step in acceptance of a steam sterilizer to confirm its readiness for validation and conformance to the URS. It establishes the initial performance capability of the sterilizer.
3) As a means to evaluate the possible effect of equipment repairs / modifications as a key component of sterilizer change control evaluation. The results would be compared to the benchmark established by its initial performance (see #2 above). If the empty chamber performance differs materially from the initial and subsequent studies, corrective measures would be necessary to return the equipment to the initially validated state. Depending upon the extent of the change being evaluated the empty chamber study could be followed by load studies.
TEST METHODOLOGY
In addition to its ill-defined origins, the empty chamber study also lacks a consensus means for execution. Its absence from regulatory standards means that operating practices and test criteria have evolved to suit the whims of those executing the studies and thus vary substantially. This publication endeavors to provide some definition of methods for performance of empty chamber studies.
Portion of the Cycle Dwell to Evaluate:
The basic expectation is to measure the temperature range across the sterilization chamber and compare it to the temperature set-point. However, evaluating the empty chamber uniformity performance is not so easily accomplished. Many sterilization cycles are specifically designed to initially target a temperature slightly above the set-point to minimize the lag time for penetration probes to reach that temperature (see Figure 1).

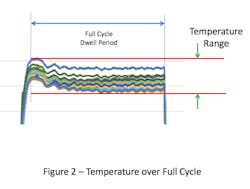
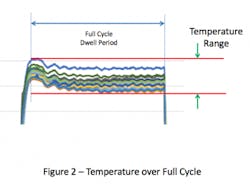
This overshoot can result in the temperatures within the chamber at the start of the cycle being higher than the set-point. If the temperature distribution within the empty chamber is evaluated over the full duration of the sterilization cycle dwell period, the range of temperature values will be greater than if it were measured any other way (see Figure 2). Inclusion of the overshot period, which is typically brief, places undue emphasis on temperatures during the initial heating, which may not have a meaningful effect on the temperature within the chamber overall.
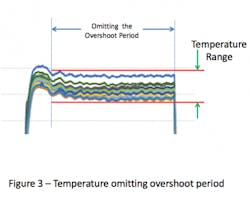
Omitting the overshoot portion of the cycle eliminates the higher temperatures observed during the beginning of the cycle and results in a smaller range of temperature (see Figure 3).
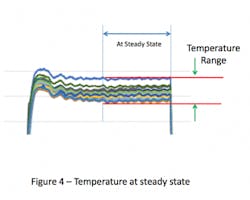
In some sterilizers (especially older models) the internal temperatures will continue to drift. If this earlier portion of the cycle dwell is omitted and the system allowed to reach steady state, then the temperature range will be even tighter (see Figure 4). Collection of temperature data over the last 3-5 minutes of the cycle has been successful for this purpose. The temperature variation over this steady state portion of the sterilization cycle can be so tight that data from a single time point might provide essentially the same result as data taken over the entire portion (see Figure 5).


The intent in showing these variations in the evaluation period dwell is to illustrate the various means that firms have elected to use to collect the empty chamber data. Collection of data over the entire cycle demonstrates the greatest variation and could be considered ‘worst case’. In contrast, collection of data at steady state is perhaps the most reproducible over time. As there are no standards for execution of empty chamber studies, there’s no fault in choosing any of these test method variations as the primary goal of the test is to establish consistency of sterilizer performance over time.
Thermocouple (TC) Locations:
It would seem obvious that measurements should be taken within the chamber, but it’s not quite that simple. At least one monitoring probe should be placed adjacent to the sterilizer control probe (usually in the sterilizer drain line) to enable easy linkage of the sterilizer data and that provided by the chamber thermocouples. Logically, if that location is outside the chamber it should not be considered in establishing the empty chamber temperature range. The empty chamber probes (and this includes the drain probe) should not be in contact with the sterilizer wall, sterilizer cart or other item.
Within the chamber, the obvious choices are the corners of the chamber near to doors, but it’s not quite that simple. The author experienced non-conforming results in one study because temperatures near the doors were higher than the sterilizer set-point! This was a consequence of having plant steam at a substantially higher pressure (and temperature) to secure the door gaskets against the chamber pressure. Other than re-piping the sterilizer and revising the control software to replace the steam with compressed air there was no ready means to remediate this variance. The actual fix was simpler, and might be considered controversial by some. The thermocouples were moved away from the corners and positioned on the corners of the sterilizer cart. This was justified because all of the load items are positioned within that cart. The added benefit to this approach was that the corner locations could be more exactly reproduced in the future.
Thermocouples could of course be placed in additional locations inside the chamber however, it seems logical to limit the locations to eight corners of the chamber as the greatest variation should be observed among those locations. Thermocouples external to the chamber in the steam supply or atmospheric vent lines can also be added, but these are merely informational and should never be included with the performance criteria.
Process Study Consideration:
The empty chamber study should be performed on a sterilizer that has been pre-heated, especially where the sterilizer is cooled for personnel access to position the temperature probes. Even where the sterilizer is not cooled, the first cycle of the day will often demonstrate a wider temperature range than later cycles. Individual empty chamber studies should be performed for cycles performed at different temperatures or using different means of heating and/or air removal. Triplicate studies are useful for new installations to assure reproducibility, but once established a single study annually (or in response to change control situations) is sufficient.
Acceptance Criteria:
As noted previously, there are no regulatory, pharmacopieal or industry documents that describe the empty chamber test in any detail, so there are no ‘official’ expectations to be satisfied. Industrial sterilizers from qualified suppliers will consistently meet the 1976 range (±0.5°C), and even better performance is commonplace for terminal sterilizers, including those of significant size (respecting of course how much of the dwell period is considered). For non-terminal usage, conformance to the same standard is clearly voluntary. As sterilization of porous loads does not require an upper limit on temperature (although many users impose an artificial one) a wider range maybe tolerated.
APPLICATION FOR OTHER STERILIZATION PROCESSES
Steam Sterilization-in-Place (SIP):
This process appears to have the greatest resemblance to an empty sterilizer chamber and thus might seem to be an excellent fit. However, the larger physical size and unique configurations of SIP systems make it difficult to have tight criteria. In the largest system with which the author has worked temperature ranges in excess of 10°C have been observed. Thus if a baseline criterion is desired, it should to be specific to the individual system. If there are a sufficient number of permanent temperature probes installed, an assessment of the system performance can be made during routine sterilization as SIP is essentially the sterilization of empty vessels.
Dry Heat Ovens for Sterilization & Depyrogenation:
This equipment is simpler than autoclaves and lends itself readily to empty chamber evaluation. The limited heat capacity of the air used makes the attainment of tight temperature ranges problematic. In small ovens with substantial insulation, a criterion of ±10-15°C might be appropriate. In larger units, and especially those installed directly on a concrete slab, the temperature range at steady state can be substantially higher approaching ±25°C or greater. Whatever the oven design or observed range, the initial operational state should be determined as the baseline for subsequent change evaluation.
Dry Heat Tunnel for Sterilization & Depyrogenation:
Tunnels which operate continuously require empty chamber assessments quite different from that of batch sterilization systems. The temperature changes from entry to exit make evaluation along the length of the tunnel of no value. Those same changes also mean that evaluation perpendicular to the belt movement can vary as well. Perhaps all that can be confirmed is the temperature range at different positions along the length of the belt which is system dependent. The initial data serves as the benchmark for change.
Non-Thermal Sterilization Processes:
Evaluation of process conditions for non-thermal sterilization methods may follow the same basic principles of mapping the interior / target area to assess the uniformity of the conditions affecting process lethality. For gas and liquid sterilization, this would mean multipoint measurement of concentration, relative humidity and temperature. Vapor (two-phase) sterilization processes rely heavily on temperature for process uniformity and should be evaluated accordingly. For radiation sterilization processes, such as electron beam and X-ray, dosimeters can be used to confirm the uniformity of the radiation dose across the target area. The acceptance criteria would be established based upon the performance after initial qualification of the equipment.
This publication has endeavored to bring greater clarity and definition to the conduct of empty chamber studies primarily for steam sterilization. The included recommendations are drawn from the author’s more than 40 years of experience with sterilization processes and must be acknowledged as opinion rather than requirements. The publication of this article may inspire some organization to establish more formal means that outlined herein.
POST-SCRIPT
This publication has focused on empty chamber distribution studies which are derived from FDA’s 1976 draft regulation which addressed loaded chamber distribution studies. In developing this publication, inclusion of content on temperature distribution was considered. It was decided that separate publication on that subject would be more appropriate. That effort is underway at the present time.
1. FDA, “Current Good Manufacturing Practices in Manufacturing, Processing, Packing or Holding of Large Volume Parenterals, and Request for Comments Regarding Small Volume Parenterals, 21 CFR 212”, Vol. 41, No. 106, pp. 22202-22219, June 1, 1976.
2. PDA,Technical Monograph, #1,”Validation of Steam Sterilization Cycles”, Philadelphia, 1978.
3. FDA, “Guidance for Industry - Submission of Documentation for Sterilization Process Validation in Applications for Human and Veterinary Drug Products”, November, 1994.
4. FDA, “Guidance for Industry - Submission of Documentation in Applications for Parametric Release of Human and Veterinary Drug Products Terminally Sterilized by Moist Heat Processes”, February 2010.
[javascriptSnippet]