Spotlight on Data Integrity
Modern pharmaceutical production lines are incredibly complex, involving international networks of raw material suppliers, manufacturing centers, testing laboratories and distribution infrastructure. Despite this complexity, it’s essential the teams responsible for releasing safe and effective products have reliable access to the up-to-date quality control (QC) data necessary to fulfill this role for each and every batch. At the end of the day, public health, consumer trust and organizational reputation depend on it.
Given the global nature of today’s drug manufacturing chains, regulatory bodies are putting the integrity of the data that underpins pharmaceutical production under ever greater scrutiny. In recent years, regulators across the world, including the U.S. Food and Drug Administration (FDA), the European Medicines Agency (EMA) and the UK Medicines and Healthcare Products Regulatory Agency (MHRA) have all issued new or updated guidance around the accuracy, consistency and completeness of pharmaceutical data, raising the bar around what’s expected when it comes to data integrity and product quality.
As a result, pharmaceutical and biotech companies both large and small are re-assessing the processes and digital tools they use to collect, handle and organize QC and manufacturing data.
Consequences of Poor data management
Many of the data integrity infringements cited by regulatory bodies are due to poor data management practices, arising from inadequate record management, incomplete documentation or a failure to adhere to validated standard operating procedures (SOPs). When it comes to taking action against manufacturers who employ unacceptable data management practices, written warnings or citations are generally the first port of call. These citations depend on the nature of the transgression but typically fall into a few main categories: properly validated systems, automatic electronic equipment, laboratory control and laboratory records.
The first type of regulatory warning, an automatic electronic equipment citation, is issued due to the improper use of digital data management systems. Examples of this include sharing unique user log-in profiles for data management systems with other individuals, failing to properly validate these systems or to establish system administrators, or the loss or deliberate deletion of data. This group of citations can also be issued if businesses have ineffective data backup processes in place.
A second type of action regulators can take is to issue laboratory control citations. These can be given if data collection does not adhere to established procedures, for instance, if individual analysts run unofficial or unauthorized tests. Preventing this type of behavior is particularly important as it can be indicative of data “cherry-picking,” where multiple datasets are collected and only the most favorable results are presented in official reports. Similarly, these citations can be issued if data is overwritten or not immediately recorded at the time of collection – conduct that can leave experimental measurements vulnerable to accidental errors or deliberate manipulation.
The third type of warning issued by regulatory bodies is known as a laboratory records citation. This can be given to a pharmaceutical or biotech company if their audit trails are incomplete or if improper review processes are in place. Given the importance of establishing a clear chain of custody when it comes to the pharmaceutical data, regulators often consider such breaches as the most egregious.
The consequences of a citation vary depending on the offense, and can range from financial penalties to being refused authorization to operate. Once organizations receive a written citation, they typically have around 15 days to respond in writing with a description of the corrective action they have taken. Any issues raised must be fixed in a timely manner, and they must not be allowed to happen again. To ensure this, regulatory authorities reserve the right to re-inspect facilities at any time.
Failure to respond to initial citations has serious implications. Warning Letters and Form 483 citations can be costly for pharmaceutical organizations, resulting in fines and even the possibility of being placed under a consent decree (a legal agreement often used by federal courts to ensure businesses comply with regulatory laws). Those that fail to respond to consent decrees can be seized and closed, and charges can be brought against the company’s leadership. Thankfully, these types of measures are often actions of last resort, with most pharmaceutical companies acting swiftly to resolve issues and implement preventative measures.
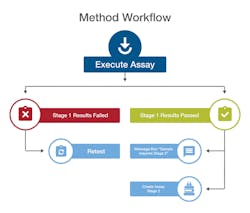
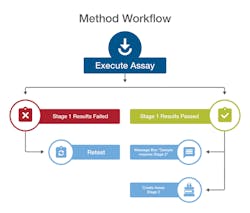
A workflow engine automatically drives samples through each stage of analytical testing to ensure compliance with methods.
Scrutinizing data management
Many of the data integrity issues highlighted arise from incomplete laboratory record management and can be avoided with the right processes and systems in place. To comply with modern regulatory requirements, laboratory records should include all of the information associated with tests and assays. This includes any data that demonstrate the function of analytical systems (such as calibration data or routine equipment checks), raw data generated during analysis, contextualized metadata, as well as audit trails. Regardless of whether paper, electronic or hybrid systems are used, these records must be accurate and kept up-to-date, and any errors will leave organizations vulnerable to citation during an audit by regulatory authorities.
To promote best practice and ensure pharmaceutical companies are “audit ready,” regulatory agencies encourage organizations to ensure their production and QC workflows adhere to the ALCOA+ principles. To achieve this, all data associated with pharmaceutical development and manufacturing should be attributable, legible, contemporaneous, original and accurate (hence the acronym ALCOA), as well as consistent, available, enduring and complete. These important principles lay the foundations for robust data integrity and outline the data collection and management practices that organizations should apply to achieve successful audits.
In addition to regulatory guidance around the ALCOA+ principles, the International Organization for Standardization’s ISO/IEC 17025:2017 requirements have also been established to help manufacturer’s QC laboratories maintain the highest standards of data integrity across all of their processes. The guidelines, which specify the general requirements for the competence, impartiality and consistent operation of testing and calibration laboratories, are not only designed to help organizations achieve the highest levels of data integrity, but also benefit from improved operational efficiency and productivity. The latest guidelines, updated in 2017, take into account the recent improvements in laboratory informatics software and digital technologies, and support the existing ISO 9001 guidance on quality management.
Multi-dimensional data integrated into a single cloud-based digital ecosystem.
Innovative laboratory informatics
The growing regulatory focus on data integrity and laboratory practices has led many pharmaceutical companies to re-evaluate the data management tools they use to organize the information handled within their QC and manufacturing workflows. With many production processes geographically disperse – sometimes spanning multiple countries and even continents – many organizations are finding that incumbent data management systems (such as lab notebooks or even spreadsheets) do not provide the level of support they need to comply with ever more stringent data integrity regulations. These platforms can leave data open to manipulation and misinterpretation, and can make recalling information for routine use or audit purposes time-consuming and resource intensive. In short, more robust solutions are required to meet modern regulatory expectations.
Increasingly, pharmaceutical and biotech companies looking to achieve the highest levels of data integrity are turning to laboratory information management systems (LIMS) to collect, organize, analyze and share their data. These powerful informatics solutions are optimizing the manufacturing and QC workflows of both large and small organizations by bringing all of their laboratory data into a single integrated system that connects instruments, people and processes. LIMS streamline data transfer, and track and manage calibration records, SOPs and experimental results. This is achieved by housing all data under one system, which is accessible and analyzable in real-time. This eliminates poor data management practices and makes demonstrating regulatory compliance to inspectors considerably easier.
One of the most notable benefits of using a LIMS to manage pharmaceutical manufacturing and QC information is the improvement in data quality that can be achieved by making the execution of workflow steps more consistent.
Take the implementation of a liquid chromatography analysis as part of a routine QC workflow, for example. Traditionally, this task would have required laboratory analysts to manually set-up sequence parameters using paper-based method details, leaving tests vulnerable to incorrect execution (such as the omission of key steps) and inconsistent updates to SOPs. By digitally storing an organization’s complete library of SOPs in a centralized hub, LIMS eliminate these issues by ensuring users have access to all of the up-to-date protocols they need to perform experiments correctly. And because assay parameters can be downloaded directly to the instrument at the click of a button, the LIMS makes sure the right experimental conditions are used for every test.
As well as making experimental set-up faster and more accurate, LIMS are also transforming the way in which QC and manufacturing chain data is collected too. This is having the most impact on processes that once relied on manual data entry, such as mass weighing or logging sample receipt. With analytical instruments and portable data collection devices (such as tablets or hand-held barcode scanners) integrated with a LIMS, all acquired data is time-stamped and stored in the system as it is generated, eliminating the need to manually transcribe or copy data from paper-based readouts or disconnected spreadsheets. Some systems can even employ on-screen prompts to guide users through data acquisition steps, and mandatory data collection fields can also be used to eliminate data omissions, increasing the quality and completeness of datasets.
With LIMS, all of this information is stored in an immediately useable and vendor-neutral format, to be used by other teams elsewhere in the production chain. This ensures that QC data used to guide batch release will be accurate, consistent and complete, and informed decisions can be made much faster.
Preserving accurate records
With regulatory bodies putting additional emphasis on the accuracy and granularity of audit trail data, another important advantage of using an integrated LIMS platform is the ease with which event logs can be generated. For many pharmaceutical companies, the knowledge that a detailed history of every event from last week, month or year can be quickly located and recalled, without having to manually trawl through fragmented or poorly managed record systems, will be particularly reassuring.
Modern LIMS platforms are able to track all user interactions, automatically capturing every addition, deletion and modification, and storing prior versions of data for long-term archiving. Furthermore, because every user accesses the system with an individual password-protected profile, authorized administrators have access to a comprehensive record of who did what, when and why from the moment the system was implemented.
Some platforms go further than this, and incorporate powerful audit trail search tools to allow administrators to quickly recall specific actions of interest. As well as simplifying the generation of reports, this granular search functionality can also make it easier to identify and investigate unusual user activity, such as interrupting sample runs or repeatedly running sample sequences. While many pharmaceutical manufacturers will hope this behavior is not a problem that concerns their own workflows, without these in-depth tools, this behavior can remain hidden until it turns into a larger problem.
With the integrity of pharmaceutical data increasingly under the microscope, growing numbers of biotech and pharmaceutical companies are turning to the most robust data management tools to achieve, and maintain, compliance. LIMS platforms bring together experimental data, SOPs, instruments, users and workflows to create a single digital ecosystem that spans the pharmaceutical manufacturing and QC pipeline. By minimizing the potential for human error, automatically collecting comprehensive audit trail data, and ensuring information is available in real time, these integrated informatics solutions can eliminate many of the poor data management practices and pitfalls that are all too commonly associated with regulatory non-compliance.