Just over a decade ago, the generics industry began enjoying remarkable success, growing rapidly and globalizing its manufacturing. This success, however, brought with it a significant regulatory challenge, and delays in generic drug approvals emerged as a major concern for the generics industry, FDA, consumers and payers, as the agency fell further and further behind in review and inspection capacities.
Signed into law in 2012, the Generic Drug User Fee Act (GDUFA) is designed to speed access to safe and effective generic drugs to the public and reduce costs to industry. The law requires industry to pay user fees to supplement the FDA’s costs of reviewing generic drug applications and inspecting facilities. Though regarded as a “historic achievement for both FDA and the generic pharmaceutical industry” by the Generic Pharmaceutical Association (GPhA), it is clear to all parties involved that the first iteration of GDUFA is not without flaws.
At the GPhA Fall Technical Conference this past October, David Gaugh, senior VP of science and regulatory affairs for GPhA, perhaps summarized it best when he said, “You don’t know what you don’t know when you start — that’s GDUFA I.”
Today, as the pharma sector continues to face sharp scrutiny in regards to pricing, more pressure is being placed on speeding the availability of low cost, high quality generic drugs. Conveniently, the legislative authority for GDUFA is expiring at the end of September 2017, so new legislation is required for FDA to continue to fund the generic drug review process for future fiscal years.
A Chance to Revisit, Revise

The GDUFA reauthorization process began with a public meeting in June 2015, which was followed by over a year of negotiations. A few of the highlights of the GDUFA II enhancements, oulined in full in the GDUFA II Commitment Letter3, include more efficient submission review goals, fairer fee structures and an enhanced pre-ANDA process for complex products.
At the GPhA Fall Technical Conference, Keith Flanagan, director of the office of generic drug policy, FDA, expressed to the packed house, “Our hope is that GDUFA II mitigates the pain points of GDUFA I.”
Backlogs
One frequently noted GDUFA issue was that of backlogs. This past January, Janet Woodcock, veteran director of the FDA’s Center for Drug Evaluation and Research, gave testimony in the Senate Health, Education, Labor and Pensions (HELP) Committee’s first oversight hearing of GDUFA.4
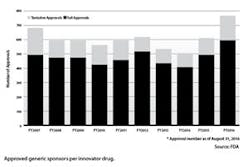
Woodcock was asked directly if the regulatory backlog of generic applications is the reason patients are experiencing higher drug costs. Since GDUFA’s launch in 2012, the FDA has had to deal with a backlog of about 6,000 ANDAs awaiting review. In her testimony, Woodcock explained that high drug prices are driven by multiple factors and yes, lack of competition is a factor. The main players here are first generics — the manufacturer receiving first approval by FDA to market a generic drug product in the United States.
Woodcock assured the committee that there were no first generics in the backlog that have not been looked at by the FDA.4 At this point, says the FDA, over 90 percent of those 6,000 applications have received some kind of review or communication from FDA officials.
According to the most recent FDA Generic Review Dashboard, 2,245 ANDAs are currently with FDA (as of October 2016) and of those applications, 1,807 have received at least one communication from agency staff and only 94 are pending review. Additionally, 1,775 ANDAs have been sent back to generic drug companies and are either pending a response from industry or have received tentative approval.
No ANDA Left Behind
In practice, the original GDUFA submission review performance goals proved to be both complex and confusing, so much so that the FDA itself called them “commercially disruptive.”
The GDUFA I review program was structured based on five cohorts of submission dates, corresponding to the five fiscal years to be covered by the program. Different cohorts and tiers of submissions received very different review goals. Some were given goal dates and some were not. This was simplified in GDUFA II, so that all ANDAs and ANDA amendments fall within a single, consolidated, review goals scheme, thus ensuring that “no ANDA is left behind.” The FDA will streamline approvals by assigning goal dates to all ANDAs.
Secondly, GDUFA II is designed to create faster review goals for priority submissions. For an ANDA, standard review will be 10 months from submission and priority review will be 8 months from submission. Priority review will be available for submissions that FDA considers to be public health priorities.
During the GPhA Fall Technical Conference, Scott Tomsky, VP of regulatory affairs generics North America for Teva Pharmaceuticals, called the revised review structure “the biggest success of GDUFA II; it’s a light at the end of the tunnel.”
New Fee Structure
Withdrawl Refunds: According to the structure of GDUFA I, if a company submits an ANDA and then decides to withdraw the submission, the company receives no refund. With GDUFA II, companies will get a 75 percent refund for submissions withdrawn voluntarily before FDA makes a decision on whether the ANDA may be received. This will save both FDA and sponsor work.
New ANDA Holder Program Fee: In order to improve the predictability of the fee base and align fee responsibility with program costs, a Holder Program Fee has been introduced. Firms that sponsor one or more approved ANDAs will pay an annual fee.
The idea behind this new fee is to “assess a fee based on a company and its affiliates every year, which will be a function of the number of approved ANDAs they have in their combined portfolios,” explained Donal Parks, director, division of user fee management and budget formulation, CDER, in a recent CDER SBIA webinar series.
The annual fee will be assessed at the beginning of the fiscal year in tiers: large (20 or more approved ANDAs), medium (6-19 approved ANDAs) and small (5 or fewer). The benefit of the tier being that those in the medium tier only pay 40 percent of the entire fee, while those in the small tier only pay 10 percent. The fee amounts are to be published two months before the fiscal year starts — August 2017.
Elimination of Supplement Fees: With GDUFA I, ANDA sponsors making changes to an already approved ANDA through a Prior Approval Supplement (PAS) were required to pay a fee. Due to the introduction of the program fee, FDA has eliminated the fee for PASs.
New CMO Fee: Contract Manufacturing Organizations (CMOs) hired by ANDA sponsors to manufacture their generic drugs will now only pay one-third of the FDF (Final Dosage Form) fee, rather than the full FDF fee. “We recognize that CMOs are most likely smaller companies, and this way the agency and negotiators wanted to address that in GDUFA II,” said Parks.
Data Clean-Up
The FDA recognizes its data may be dated or inaccurate, and seeks to clean up its ANDA applicants/sponsors data in advance of implementing new ANDA Holder fee for GDUFA II. Starting this December, the FDA will post an Excel file list of all the approved ANDAs, grouped by the name of the holder of record. Multiple company names will equal multiple fees. The agency is giving companies the opportunity to correct any errors. The FDA will then republish an edited list in March, reflecting corrections and tiers, and publish a final list in June 2017.
Recognizing Complex Products
“Complex products” are now a defined term in the GDUFA II Commitment Letter. According to the FDA, complex products are “products with complex active ingredients, formulations, routes of delivery or dosage forms; complex drug-device combination products; and other products where complexity or uncertainty concerning the approval pathway or possible alternative approach would benefit from early scientific engagement.” Peptides, Liposomes and transdermal systems, for example, all fall into the bucket of complex products.
Complex products pose distinct scientific and regulatory challenges. The prevailing sentiment at the most recent GPhA Technical Conference surrounding complex products was that of confusion - applicants didn’t know what FDA expected - making it much harder to develop approvable ANDA for submission. The agency, as well, has expressed frustration in regards to receiving incomplete submissions in general. In Janet Woodcock’s January 2016 testimony before the Senate Committee on Health Labor and Pensions, she stated that ANDA submission quality is an ongoing challenge, noting that historically it has taken on average about four review cycles to approve an ANDA as a result of deficiencies by generic drug sponsors in submitting complete and quality applications.
In an analysis of GDUFA I and ANDA operations at the GPhA Fall Technical Conference, Ted Sherwood, director, office of regulatory operations, OGD, FDA, reminded the audience that, “Your ANDA is trying to tell a story, and it’s important that it tells the story that the agency needs to hear.”
Accordingly, new to GDUFA II is a pre-ANDA program for complex products. The goal of this upgrade is to clarify regulatory expectations for prospective applicants earlier in product development and help applicants develop more complete submissions, therefore reducing the number of review cycles to obtain ANDA approval of complex products.
GDUFA II’s pre-ANDA program aims “front-load” work so ANDAs can be right the first time.
The GDUFA II Commitment Letter includes a discussion of product development, pre-submission and mid-review cycle meetings for complex products, as well as clearly defined parameters for all meetings. Additionally, the FDA has said that it would strive to issue product-specific guidance for complex products as soon as scientific recommendations are available.
Ensuring Generics’ Future
According to the 2016 Generic Drug Savings and Access in the United States report, generics make up 89 percent of prescriptions dispensed in the United States, but consume just 27 percent of the total drug spending. Generic drugs resulted in $227 billion in savings in 2015.
As generic competition continues to play a key role in driving access and cost savings, it is important to continue building on the foundation laid by GDUFA I. The five-year GDUFA II program will provide the FDA with $2.6 billion in financial resources to help ensure Americans continue to receive timely access to safe, effective and affordable generic drugs.
Though ongoing and thorough negotiations and conversations with industry, the FDA has recognized an overall need to be more specific and programmatic and step up the communication and transparency on multiple levels. The resulting second iteration of GDUFA looks to provide a clearer pathway for future generics.
References
- Generic Pharmaceutical Association. (2014). Statement from Ralph G. Neas, president and CEO, GPhA [Press release].
- The U.S. Food and Drug Administration, Department of Health and Human Services. (2016). Generic Drug User Fees; Public Meeting; Request for Comments
- The U.S. Food and Drug Administration. (2016). GDUFA II Commitment Letter. Retrieved from http://www.fda.gov/downloads/forindustry/userfees/genericdruguserfees/ucm525234.pdf
- U.S. Senate Committee on Health, Education, Labor and Pensions. (2016). Full Committee Hearing: Generic Drug User Fee Amendments: Accelerating Patient Access to Generic Drugs
- The U.S. Food and Drug Administration. (2016). The Generic Drug Review Dashboard Quarterly Update
- The U.S. Food and Drug Administration. (2016). CDER Small Business and Industry Assistance Webinar - Overview of GDUFA II and Implementation of GDUFA II User Fees
- Generic Pharmaceutical Association. (2016). 2016 Generic Drug Savings and Access in the United States Report
About the Author
Karen P. Langhauser
Chief Content Director, Pharma Manufacturing
Karen currently serves as Pharma Manufacturing's chief content director.
Now having dedicated her entire career to b2b journalism, Karen got her start writing for Food Manufacturing magazine. She made the decision to trade food for drugs in 2013, when she joined Putman Media as the digital content manager for Pharma Manufacturing, later taking the helm on the brand in 2016.
As an award-winning journalist with 20+ years experience writing in the manufacturing space, Karen passionately believes that b2b content does not have to suck. As the content director, her ongoing mission has been to keep Pharma Manufacturing's editorial look, tone and content fresh and accessible.
Karen graduated with honors from Bucknell University, where she majored in English and played Division 1 softball for the Bison. Happily living in NJ's famed Asbury Park, Karen is a retired Garden State Rollergirl, known to the roller derby community as the 'Predator-in-Chief.'