Risk Management for Regenerative Medicine Distribution

Regenerative medicine will soon be the standard of care for replacing tissue/organ systems in the human body, according to the United States Department of Health and Human Services Report, “2020: A New Vision—A Future for Regenerative Medicine.” The impact of this shift will be huge. For instance, a definitive cure for heart-valve disease in the U.S. alone would provide an annual cost savings of $23.4 billion.
Reaching that potential has been anything but a simple task. Manufacturing cell-based therapies is expensive, and companies have faced considerable challenges to achieve standardized quality using methods that are scalable and sustainable.
Still, all of that expense and effort will be for nothing if the product is damaged or destroyed while in transit. Protecting the product in the last mile is every bit as important as how it is protected during every other step.
The Need for Updating
The emergence of regenerative medicine as a viable therapy class has amplified the focus on current clinical product distribution standards and the need for enhanced requirements that parallel current manufacturing standards in the industry. The regenerative medicines market generated $17.03 billion in revenue in 2016 and is expected to reach $50.55 billion by 2025.
The fragility of RMAT products is not what past supply chains were built for. The industry is seeing accelerating clinical progression for personalized therapies for diseases from hemophilia to cancer in as little as four years. This timeline compression has placed enormous pressure on existing supply chains, which were never set up to effectively manage risk for a drug product that is modified for a single individual — and in most cases does not have back-up doses if something goes wrong during transport or storage.
Clearly, the industry faces an urgent need for new clinical product distribution and logistics standards as robust as the standards for manufacturing.
One common observation is that current cold-chain container qualification and management processes are insufficient in effectively managing risk during personalized medicine distribution. Basic requirements outlined in 21 CFR 210 and 21 CFR 211 for traceability of equipment utilized in the manufacture and storage of drug substance and drug product must be implemented and utilized in the drug distribution space.
To that end, both the Alliance for Regenerative Medicine (ARM) and the Foundation for the Accreditation for Cellular Therapy (FACT) have instituted review bodies to look at all aspects of the collection, manufacture, transportation, and administration of regenerative medicines.
Moreover, emerging Chain of Compliance requirements for regenerative medicine distribution are rapidly becoming the standard for ensuring product integrity.
The Chain of Compliance
Early on, the industry correctly understood traceability as links in a chain:
- Chain of Custody: Traceability of the custody of each client’s or patient’s therapy
- Chain of Condition: Traceability of the condition of each client’s or patient’s therapy
- Chain of Identity: Traceability of the identity of each client’s or patient’s therapy
But the fragility of RMAT products — and the urgent need for standardization — means that a fourth chain is now required: the Chain of Compliance.
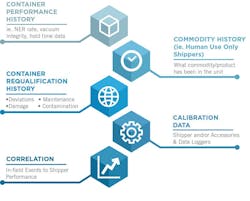
This traceability can be addressed using Chain of Compliance processes which effectively manage this traceability. Chain of Compliance is establishing full traceability of the equipment and processes used in managing the environmental control of the commodity. This includes container performance and requalification history, commodity history, courier handling and performance history, calibration history, and correlation competencies that can link in field events to equipment performance. A review of these requirements are as follows:
- Container performance history: All transportation equipment has a validated hold-time standard that can change over time for multi-use equipment. Data supporting an accurate calculation of the hold-time of a cold chain container might include the nitrogen evaporation rate, liquid nitrogen capacity, vacuum integrity, dynamic hold time, as well as the actual in field temperature, humidity, shock and orientation data.
- Commodity history: In addition to the performance of the equipment utilized for a given shipment, a complete historical ledger of the contents shipped in any given container should be tracked so that one has the ability to determine if a given piece of equipment has only ever been used for the distribution of non-infectious human materials and can certify this history.
- Container (re)qualification history: Additionally, one needs to have accurate records as to the requalification or testing of the performance of the equipment to be utilized. These records should also include any repairs or maintenance done to the equipment, any deviations or damage during use, as well as any contamination or sterility issues over the entire historical usage of the equipment in question.
- Calibration history: All calibration data for any electronic components of a given package must also be traceable back to such equipment. This would include thermocouple calibration or validation data, battery performance, software or firmware updates by date and version, and serialized accessories that are archived by part number.
- Correlation: Lastly, the ability to cross reference field handling events including shock, damage, delays, orientation and anti-tamper competencies to the impact on the commodity shipped is a key requirement. Additionally, one should be able to cross reference the historical custody of the tank as well. This would include all locations receiving the tank, as well as the courier for freight partners who were responsible for the delivery of the tank from origin to destination.
Complete Data
The reason that the FDA and other regulatory bodies are interested in Chain of Compliance is that it provides the ability to collect, interpret, and leverage comprehensive data, enabling a significantly more intelligent supply chain. Rather than reactively trying to determine what has gone wrong after multiple failures, it becomes possible to take a proactive approach. Moreover, effective implementation provides historical traceability of logistics processes, equipment and third party support entities, enabling one to critically assess its complete supply chain and minimize failures and risk.
What’s more, having complete data gives you the ability to significantly reduce the risk of product failure. Robust data reveals patterns you can leverage year after year.
With a complete picture of every element and a data-rich understanding of how those work in the real world, you can apply Quality by Design risk management processes to your supply chain strategy. QbD is a systematic approach to development that begins with predefined objectives and emphasizes product and process understanding and process control based on sound sciences and quality risk management. Put simply, QbD yields better results than “Quality by Hindsight.”
A Strong RMAT Supply Chain in Action
What should companies demand from a modernized, RMAT-ready supply chain?
Obviously, there will be individual company-level differences but at a baseline level the following are necessary.
Unit Level Performance
This approach includes:
Serialized packaging that tracks the performance of each unit.
All equipment managed by a QbD process that incorporates risk due to transit related events.
Intelligent unit level tracking prevents commissioning of equipment that does not have requisite specification for the lane/commodity selected.
“Fuzzy Logic” Risk Management
A robust informatics package should provide:
- Proactive alerts (it’s no good to open the dewar on arrival only to discoverer it has not been maintained at the correct temperature). Also, when alerts occur there should be clear, standardized SOPs for escalation: People need to know what to do when there’s a problem.
- A method for correlating transit-related events to product integrity.
- Constant monitoring for deviations, exceptions and other handling events.
- A method to maintain historical carrier performance by lane and packaging.
Reporting
All reporting should:
- Be 21 CFR Part 11 compliant.
- Have a database that retains all transit and packaging records.
- Be capable of providing certification and performance reporting at the unit level.
Regulation is Racing to Catch Up
The pace of change in regenerative medicine can seem daunting. It demands a balance of speed and proactive management while carefully guarding against downside risk. Yet the promise to dramatically improve patient outcomes and create important new lines of business means there has never been a more exciting time to be part of this industry.
It’s likely that regulations from the FDA will be established in the next 12 months. Not all companies will be ready to respond, but those that do will be positioned to leapfrog past competitors who take a wait-and-see approach.
Regenerative medicine is already revolutionizing the treatment of diseases, and promises to rapidly scale in the coming quarters, putting additional stress on supply chains used to distribute therapies that have no tolerance to excursions. Reconsidering or completely re-engineering your cold supply chain to support future regulation is entirely necessary — and worth the effort.
- Continuously track individual equipment performance.
- Track and archive what’s being put into your equipment and who is moving it.
- Ensure you have comprehensive (re)qualification and maintenance records for your equipment.
- Integrate all four chains — custody, condition, identity, and compliance — into a single data stream for cross-referencing and accountability.
- Insist on world-class informatics, and real-time integrity measurements.
[javascriptSnippet]