Grasping Lyophilization Product and Process Parameters for Biopharma
Editors Note: This is an excerpt from Chapter 3 of Dr. Franks latest book, Freeze Drying of Pharmaceutical and Biopharmaceuticals: Principles and Practices, published last year by the Royal Society of Chemistry (copyright reserved). Dr. Franks, founder and director of the BioUpdate Foundation, has long been an advocate of understanding the physical, chemical and biological properties involved in freeze drying. His early research focused on the vitrification of aqueous media, which led to the development of new ways to stabilize biomolecules in vitro. He established Pafra Biopreservation to develop these processes commercially and apply principles of materials science to the process.
Nektar Therapeutics later applied that know-how in the inhalable insulin drug form that would become Exubera. In his book, Dr. Franks recalls being invited to help a company specializing in human blood derivatives, which had been achieving highly variable results in its freezedrying processes. He writes: During a roundtable discussion, it became apparent that those pharmaceutical chemists who were responsible for formulation development had never met or spoken to their engineering colleagues, who were responsible for the freeze-drying process. Indeed, for security reasons, the latter were not informed of the nature of the products that were to be freeze-dried. This was a scenario that our technologists also encountered in a number of other mega-pharma companies. Usually, even the mention of glass transition or its measured value met with blank stares.
The complexities were not appreciated, the freeze-drying operation being regarded as a push-button affair. hardly a recipe for success Later in his book, Dr. Franks discusses some case studies with specific lyophilization processes. For one bioactive substance, a client found that the process stopped working entirely. It was later discovered that the company had changed to a different set of narrower vials, in order to increase loading capacity, but they did not take into account the increased fill depth. Dr. Franks writes, Process development staff [often] knew little or nothing of glass transitions or the materials science approaches to the formulation of amorphous pharmaceutical solids. Even where the knowledge exists within a company, its significance does not generally reach the thinking of marketing or senior management. Too often the author has been invited by departments to make presentations to senior managers, with the sole purpose of convincing them of modern thinking on freezedrying. Unless this lack is remedied, freeze-drying will continue to be practiced as a trial-and-error technique. Chapter 3 presents an overview of the basic product and process parameters essential to biopharmaceutical freeze drying. To order the book, visit www.rsc.org/shop/books.
The physics and chemistry of freezing show many complex and often surprising features, but the industrial freeze-drying process is limited to very few operational degrees of freedom. Thus, for a given solution composition and a given container and closure system, only three process variables exist by which direct control over the drying cycle can be maintained and which, therefore, determine the quality of the final dried product. They are:
- shelf temperature
- chamber pressure
- time
On the other hand, the most important factor is of course the product temperature and its change throughout the duration of the process.
Formulation Parameters
In the manufacture of a therapeutic product, the initial stage requires the purification of the biologically active component, the drug substance. Purification methods for conventional drugs are well established. They are based on synthetic organic chemistry and standard analytical techniques.
The situation is more complex for biopharmaceutical products, in particular those based on proteins. The drug substance may then be of animal, plant or microbial origin; it may be obtained from the natural source materials or by recombinant DNA (rDNA) methods. The extraction and purification strategy depends to some extent on the source of the protein. Possible impurities are many. The downstream processing always requires several stages and, depending on the source material, may involve a combination of physical and chemical methods.
The total yield of purified protein is related to the number of stages required and their individual yields. For instance, in an isolation and purification process consisting of n steps, each one of which can be carried out with a 90% efficiency (highly idealised!), the final percentage yield of product is given by:
Yield (%) = 9n x 10(2-n)
Thus, for a process consisting of five stages, each of which can be performed with a 90% recovery, the yield of the final product cannot exceed 60%, which is reduced to 53% for a six-stage purification procedure. Since biopharmaceutical drug substances, whatever their origin, are usually expensive, a loss of 40% during the purification process is a serious factor. A minimization of the number of stages is therefore aimed at, but it has to be judged against other economic considerations and an acceptable degree of purity.
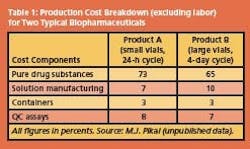
For biochemical and therapeutic uses, purity and longterm stability are the overriding requirements. Fractionation, purification and stabilization by freeze-drying then generally account for 50% of the total production costs, but since such products command high premiums in the marketplace, production hardly figures in the cost equation. Examples of typical production cost breakdowns for two product/process scenarios are shown in Table 1.
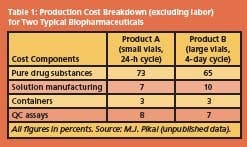
The data illustrate that the cost contribution allocated to freezedrying is almost insignificant, compared to the cost of the purified raw drug substance, assumed to be a protein. Formulation considerations Before a purified protein, usually in a dilute aqueous solution, is subjected to the final stabilization procedure, e.g. by freeze-drying, it has to undergo a process of compounding, loosely referred to as formulation.
Very few proteins can survive freeze-drying without the aid of the so-called lyoprotectant additives, which serve to ensure the recovery of full biological activity of the protein at the point of use, whether in the dry solid state or reconstituted in an aqueous medium. The science and technology of lyoprotection, as employed in freeze-drying, is only gradually being put on a quantitative basis; several unresolved mysteries remain.
Apart from lyoprotectants, other substances are also commonly added to the protein solution prior to drying. Some may also have been carried over in the solution during downstream processing. They may include pH buffers, surfactants and salts. A common additive, phosphate-buffered saline solution (PBS) ensures the isotonicity of a reconstituted parenteral product in water, prior to injection. Although from a pharmacokinetic standpoint some of the additives in common use may be innocuous or even beneficial, their presence in the solution may increase technical problems that can be encountered during freeze drying.
For instance, the presence of PBS will inevitably require a longer freeze-drying cycle than would be required for the same solution in the absence of salts. The omission of phosphates and NaCl from the initial solution is therefore helpful, since it allows for a shorter drying cycle. On the other hand, reconstitution must then be performed with PBS solution, rather than with sterile water (water for injection), which apparently is a distinct disadvantage from a marketing standpoint.
Process Parameters
As already pointed out, the primary process parameters by means of which drying can be directly controlled are shelf temperature, chamber pressure and time. In principle, the condenser temperature can also be adjusted, but in practice it is usually set to the lowest possible value, so as to maximize the ice sublimation rate. Since it is the temperature of the material that determines the drying rate, the control over the process can only be indirectly maintained, usually by coupled adjustments, either continuous or ramped, of the shelf temperature and/or the chamber pressure throughout the drying cycle.
Once the process cycle conditions have been decided upon and the vials have been loaded, the process is automatically controlled by a computer. The freeze-dryer performance is monitored, and the time course of the production cycle (48 hours in this case) is recorded in terms of shelf, condenser and product temperatures and chamber pressure. As mentioned earlier, the only directly adjustable parameters are the shelf and condenser temperatures, and the chamber pressure. We shall defer a detailed discussion of this information to several later chapters.
Understanding Freeze-Drying
The successful application of freeze-drying is based on a complex interplay of several scientific principles, some of which are still very poorly explored and hardly appear in university undergraduate curricula. In the authors experience, one rarely finds managers or technologists with an overall broad view of the various stages of freeze-drying. Thus, the trained chemists experience differs from that of the physicist, the materials scientist or the chemical engineer. And yet, an overall appreciation of how these disciplines interact is of the utmost importance.
Experts in the various disciplines within a company will usually be found in different divisions of a line management structure, a custom not conducive to the fostering of the necessary collaboration. Key disciplines include: Pharmacokinetics deals with all aspects of the therapeutic effect of the chosen drug substance, suitably formulated, on its target. It forms the very basis of the formulation and processing stages. Formulation. In the case of biopharmaceuticals based on proteins, some experience of protein biochemistry and technology is absolutely necessary.
The formulation process aims to protect the bioactive substance from the severe stresses to which it will be exposed during any drying process, including those directly due to concentration increases during freezing. The freezing process is governed by ice nucleation and crystal growth rates in supersaturated solutions. It is accompanied by freeze concentration of the solution, a process that may result in both crystalline or amorphous solids, or in mixtures.
Materials Science. Correct identification of the physical and chemical identities of the product during and after processing requires an understanding of the analytical techniques available for obtaining the necessary information, and for eventual submission to the relevant regulatory body. The ice sublimation stage depends on the balance of heat and mass transfer. Secondary drying depends on the diffusion rate of water through the solid matrix. The long-term stability is governed by the materials science of crystalline and amorphous solids, and it is measured with the aid of an array of physical analytical techniques, such as thermal analysis, X-ray diffraction, spectroscopy and chromatography.
For registration purposes, all process and analytical data must be submitted in the required formats, with product identity, purity, safety, stability and shelf life playing major roles in the submission for regulatory approval.
About the Author
Felix Franks is founder and director of the BioUpdate Foundation. He has worked as systems consultant to Nektar Therapeutic Systems, and has been a Special Professor of Life Sciences at the University of Nottingham (UK) since 1973.