A Case for Stage 3 Continued Process Verification

FDA Process Validation Guidance (Guidance for Industry: Process Validation- General Principles and Practices, Jan. 2011) outlines process validation activities in three stages - Stage 1: Process Design, Stage 2: Process Qualification and Stage 3: Continued Process Verification. Completion of Stage 2 subsequent to Stage 1 is a major milestone in the Process Validation Lifecycle as it confirms the process design and demonstrates the expected consistent performance of the manufacturing process. Knowledge and information gained from the design stage through the process qualification stage is used to complete this assessment. Stage 2 demonstrates suitability for successful commercial distribution where the data indicates that the process meets the conditions established in the protocol. Continued Process Verification is initiated for the subsequent commercial batches. Stage 3 assures that the process remains in a state of control during commercial manufacture.
21 CFR 211.180(e) regulations require evaluating and determining the need for change in manufacturing or control procedures on an ongoing basis. ICH Q8 (Rev 2) recommends an enhanced Quality by Design (QbD) approach that is comprised of a process validation lifecycle with a process verification stage. The March 2012 EMA Guidance on Process Validation requires continued process verification during commercial manufacture. This ensures a continued state of process control throughout commercial production.
Implementing Stage 3-Continued Process Verification makes business sense as it allows for freeing up of production lines and a higher throughput of the manufacturer’s portfolio. The ultimate result of a well-implemented Process Verification Process is an uninterrupted product supply that provides enhanced client service levels.
COMMERCIAL PRODUCT LAUNCH
As recommended by FDA and EMA (QbD), a design space is established at early stages of product development based on experiments conducted at pilot scale. The commercial process is operated in a specific area of the design space, i.e., Normal Operating Range (NOR), close to target operating conditions. NOR for a commercial process is defined upon evaluation of potential scale-up effects. Continuous Process Verification is to occur within the NOR. The continuous monitoring plan for quality attributes and/or process parameters as part of the routine control strategy will further strengthen a QbD-based product submission.

Example Section of an FMEA Model Risk Assessment
Stage 1, Process Design regulatory expectations are clear for a commercial product launch. The FDA’s Compliance Policy Guide (CPG) Sec. 490.100 - Process Validation Requirements for Drug Products and Active Pharmaceutical Ingredients Subject to Pre-Market Approval - requires proof of process validation be obtained through rational experimental design and systematic evaluation of data, beginning from the process development phase and continuing through the commercial production phase.
Preliminary Risk Assessment and multivariate design of experiment (DOE) studies help to reveal manufacturing relationships and interactions and to establish operating ranges. These findings are documented in the product development report. For a typical solid dose manufacturing process, information available from the product development stage is leveraged in the technical transfer/scale-up studies. A technical risk evaluation (see Figure 1) prior to technical transfer/scale-up studies for solid dose products include elements such as:
- Review of API characteristics (solubility, polymorphism, hygroscopicity, particle size)
- Critical excipient characteristics (ex. MCC)
- Dissolution profile (ex. immediate release, modified release)
- Formula (API%, use of well-established excipients, etc.)
- Equipment and tooling (ex. new press, tablet shape)
- Manufacturing process (powder flow, new mfg. technology)
- Product development batches and parameter range studies (ex. blend time studies, Compression Specification Range Determination study, coating study)
- Bulk hold time results
- Evaluation of CPP
- Specifications
- Stability and stress studies
- Packaging configuration
A Risk Priority Number (RPN) is calculated based on development data, process understanding and historical performance data for similar products. The continuous monitoring program is a prime resource for gathering historical process performance data. The data from similar product/process can be used for a probability risk assessment.
BENEFITS FOR NEW PRODUCTS
Stage 2 Validation study batches are targeted at substantiating the parameters defined during the Stage 1 - Process Design (process development, ranging studies and scale up studies), where the commercial manufacturing process is defined. Conformance batches (Stage 2 - Process Qualification) are prepared to demonstrate that under “normal conditions and operating parameters” the process produces an acceptable product. Post commercial launch monitoring is critical for new products where there is limited product data or experience. Though the FDA guidance (Guidance for Industry: Process Validation - General Principles and Practices dated Jan. 2011) does not require splitting of Stage 3, a specific Continued Process Verification stage involving a statistically relevant number of batches to gather adequate trending data is highly recommended for new commercially launched products where historical product data is not available. For the sake of convenience, this new stage is referred to as “Stage 3A.”
Stage 3A is designed to allow close monitoring of parameters and quality attributes and to detect any undesirable process variability trends observed post launch, thus providing an opportunity to make recommendations and finalize the routine Continued Process Verification (Stage 3B) control limits. Stage 3A is used to perform additional sampling and testing (monitoring), if required, based on the Stage 2 assessment. Stage 3A is performed under a pre-approved protocol. Stage 3A Continued Process Verification Protocol uses statistical measures to determine process performance. Process performance (Ppk) values are computed based on a predetermined number of consecutive commercial batches. Ppk values and Statistical Process Control (SPC) charts are developed for in-process critical quality attributes (CQA) and critical process parameters (CPP) where applicable. Similarly, process performance capabilities and probability of Dissolution Acceptance (Pa) are determined for the Finished Product critical quality attributes. A Stage 3A Report includes a discussion around any excessive intra-batch variability.
Accepted statistical guidelines indicate processes with Ppk more than 1.0 are within statistical control. Appropriate actions (changes, continuous improvement activities, etc.) are discussed in the report if Ppk values are less than 1.0. The Stage 3A statistical analysis provides a science and risk-based assessment of the product meeting the critical quality attributes in the future.
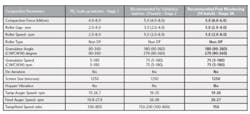
Typical Representation of Solid Dose Compaction Parameters
Table 2 shows how compaction parameters might be adjusted post Stage 3A to consistently meet the established quality attributes. The continued batch to batch verification at Stage 3B will further highlight any undesired trends.
RE-VALIDATION
A decision to re-validate (Stage 1 and Stage 2) the commercialized manufacturing process starts with the change control process. Supporting data for the change and a technical risk assessment document the process re-validation strategy. Examples of process changes where re-validation may be recommended are:
- Investigation/failure related change recommendations
- Formulation and/or process changes such as scale up/scale down
- Continuous process improvement recommendations
- Material and/or equipment changes
- Regulatory notifications, etc.
Batches are not released for commercial sale until Stage 2 is successfully completed for the change (Refer to Figure 3 - Stage 1, 2, 3, Changes).
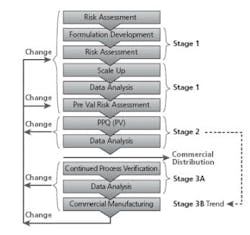
PV Lifecycle (Stage 1, 2, 3)
Continued Process Verification (Stage 3B) trend reports provide a statistically relevant and current capability score. This information is used for deciding a validation strategy. For example, a lower dissolution probability (low probability of meeting the USP Stage 1 dissolution criterion) may warrant more batches and additional sampling at Stage 2 re-validation to ensure the change did not adversely impact dissolution results.
Figure 4 displays a statistical model developed to determine the minimum number of batches required for Stage 2 re-validation on the basis of the products current capability score. Stage 3B is used to establish a scientifically justified sampling and testing plan while performing the risk assessment and drafting of the Stage 2 re-validation protocol. A review of Stage 3B trend reports aid in gaining knowledge and performance insights of similar process/products. This data is relevant in continuous improvement and determining optimal processing parameters through product development and scale-up activities.
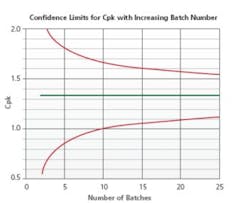
Number of Batches Required
FDA Guidance Process Validation: General Principles and Practices (P. 11) recommend use of credible experience with sufficiently similar products and processes. The cumulative data from all relevant studies (e.g., designed experiments; laboratory, pilot and commercial batches) are used to establish Stage 2 requirements. Bracketing of multiple strengths may be possible with an assessment that demonstrates low risk due to the change based on the Stage 3B trend. A possible candidate for such an approach is wherein an API change has a minor impact to the process and there is very solid Stage 3B trending. A bracketing approach to Stage 2 re-validation, such as three batches of the high and low strengths and one each of the middle strengths, may also be employed.
Another measure of interest, especially during development of a re-validation strategy, is the Stage 3B capability score for routine blend and dosage uniformity results. A low score may indicate a potential segregation concern. The Stage 2 re-validation sampling and testing plan must be developed in a manner that, though low, mitigates the identified risk. A risk-based (stratified beginning and end) sampling plan and an example outcome is provided in Figure 5.

Risk based stratified sampling plan
BATCH-TO-BATCH TRENDING
Stage 3B control limits (UCL and LCL) are developed based on statistical analysis of historical product data (Stage 1, Stage 2 and Stage 3A) for trending of the CPPs and CQAs. These limits are tighter and different from the in-process/finished product Out Of Trend/Out Of Specification (OOT/OOS) criteria developed. Any recommendation to update applicable master/specifications/test profile with the Stage 3B limits may be provided in the Stage 3A report. Under most protocols, an OOT or OOS result automatically triggers an investigation, however deviation from Stage 3B limits would not necessarily trigger an immediate investigation. Instead it may require either: 1) a scientific rationale (that justifies minimal or no impact to patient safety and efficacy) for continuing with the process; 2) initiation of a change request or 3) commencement of a continuous improvement remediation project.
Additional periodic re-assessment of the validated status of a process is not relevant due to the continued process verification Stage 3B, where this is being assessed on an ongoing basis by an experienced process analyst. However, an Annual Product Review, which is a holistic review of the product encompassing review of complaint trends, field alerts, etc. is still required. Stage 3B continued process verification may begin after Stage 2 without undergoing Stage 3A in cases where data are sufficient and risk is low. The trending tools can be defined in the Stage 3B procedure and the acceptance criteria defined in Stage 3B specifications. A product specific protocol may not be required for the continued process verification stage 3B.
The Stage 3B trend report is comprised of documented evidence of CPPs, in-process and finished product CQA trend plots, as well as their analysis and recommendations. Some of the attributes/parameters that may be trended during Stage 3B for a solid dosage manufacturing (see Table).
Instances where the product data appear to trend in an adverse manner are evaluated in more detail. Stage 3B is part of the PV Lifecycle and only terminates upon product discontinuation. This stage potentially collects the most comprehensive process data for the product during its lifecycle (i.e., Stage 1, 2 and 3). Existing validated manufacturing processes with a large amount of product/process data, that have undergone multiple annual product reviews, may be placed directly at the 3B Continued Process Verification stage.
There are multiple qualified automated solutions available for implementing the Stage 3B continued process verification. The primary requirement is to identify the data sources depending on manufacturing solutions (e.g. SAP) implemented at the manufacturing site. Due to the high resource requirement, time and potential for data error, a manual data collection process is not recommended for stage 3B. Stage 3B typically requires powerful statistical software for capability trending and building statistical models. FDA Guidance for Industry: Process Validation- General Principles and Practices (P. 14), recommends that a statistician or person with adequate training in statistical process control techniques develop the data collection plan and statistical methods for procedures used in measuring and evaluating process stability and process capability (Stage 3B). The FDA Guidance further recommends having procedures for guarding against overreaction to individual events as well as for failure to detect unintended process variability.
ADVANTAGES OF IMPLEMENTATION
Dr. Pradeep Sanghvi, executive vice president, Global R&D, Apotex Inc. highlights the significance of Stage 3 validation by noting that “the continued monitoring phase proposed by FDA is the logical enhancement of the Quality by Design approach.” A QbD based approach as part of Stage 1 (Process Design) works in tandem with the principles illustrated in the FDA PV Guidance, EMA Guidance on PV, and ICH Q8, 9 and 10. The agencies encourage firms to adopt innovative approaches such as Process Analytical Technology (PAT) wherever applicable with a goal to reduce process variability that may impact the CQA’s. Stage 3-Continued Process Verification is an effective quality risk management tool for detecting trends and implementing preventive measures prior to CQA failures.
As outlined above, implementing Stage 3-Continued Process Verification and the elimination of process related failures makes business sense due to the resulting:
- Reduced cost of quality.
- Improved process control and process understanding.
- Continuous supply chain which will enhance client service levels.
- Better manufacturing planning.
- Possibility for addition of more products to the manufacturer’s portfolio.
- Reduced regulatory and compliance risk.
Regulatory agencies such as U.S. FDA expect positive outcomes from these novel initiatives. Their commitment to the guidance is reflected in recent Warning Letters where excerpts from the 2011 Guidance are referenced. Being pharmaceutical professionals serving a critical patient population, we must ensure that we utilize data from all stages including Stage 3 (Continued Process Verification) and employ scientific methods, principles to shape the foundation of our conclusions.
About the Authors:
Daniel Alsmeyer is currently Scientific Leader, Statistical Support at Apotex Inc. the largest Canadian-owned pharmaceutical company. He received his PhD in Analytical Chemistry from The Ohio State University and holds multiple patents related to PAT. He has previously led analytical labs for a major chemical company and has also worked as an independent industrial statistics consultant in the United States. He can be reached at [email protected].
Ajay Pazhayattil M. Pharm is currently Technical Operations-Process Validation Manager at Apotex Inc. He is a certified GMP professional who has previously lead Validation, QA and Compliance groups at major Pharmaceutical brand name, generic and contract manufacturing organizations in North America and India. He can be reached at [email protected]
References:
FDA Guidance for Industry: Process Validation- General Principles and Practices (2011).
EMA Draft Guideline on Process Validation (2012).
ICH Q8 (R2), 9 and 10.
FDA Guidance for Industry PAT.
FDA Questions and Answers on Level 2 Guidance- Production and Process Controls.
Conclusions of FDA-EMA Parallel Assessment of Quality-By-Design Elements.
FDA Compliance Policy Guide (CPG)- Process Validation Requirements for Drug Products and Active Pharmaceutical Ingredients Subject to Pre-Market Approval.