US Pharmacopeia General Chapter <231> Heavy Metals, which describes a wet chemistry procedure for the analysis of heavy metal elemental impurities in pharmaceuticals that was developed more than 100 years ago, has finally been updated as USP <232> and USP <233>. However, there is still some confusion within the pharmaceutical industry about who is responsible for performing elemental impurity analysis and there are concerns and questions over which products should be tested: does the final drug manufacturer have to test the finished product? Do excipient manufacturers have to supply drug manufacturers with the appropriate data? The reason for the confusion is that the USP has published a number of different options regarding the analysis of elemental impurities.
IDENTIFICATION OF DRUG CONTAMINANTS
The purpose of elemental analysis of pharmaceuticals is to identify impurities that may contaminate pharmaceutical products. To ensure that all pharmaceutical product components and their manufacturing procedures comply with the regulations, risk assessment is now a priority for all pharmaceutical manufacturers. This however, can pose quite a challenge for manufacturers, especially when all the potential sources of impurities are considered. Such sources include excipients, water, the active pharmaceutical ingredients themselves, as well as container systems and manufacturing processes. When a potential risk is identified, additional data are required and testing for elemental impurities becomes the next challenge.
THE SUMMATION APPROACH
The summation option proposes that the amount of each elemental impurity present in each of the components of the drug product be determined and that the total amount of each impurity present be compared with the daily PDE limit for each species. While this is a costly tactic, it does give a higher level of control. Manufacturers can monitor individual components before they are introduced into the manufacturing process, and production can be stopped if the amount of an ingredient exceeds allowed limits. In using the summation approach, the manufacturer must also ensure that additional elemental impurities are not added to the product during the production process and that the container-closure system used is not a potential source of contaminants. If the total amounts of impurities from each of the individual components are within the specified limits, the final drug product may not need to be tested: hence the name ‘the summation approach’. It is important to note however, that the validation for this analysis must be quantitative, and that it is necessary to determine whether the manufacturing process or the drug container-closure system being used could add any elemental impurities to the drug.
ASSESSING THE RISK OF CONTAMINATION
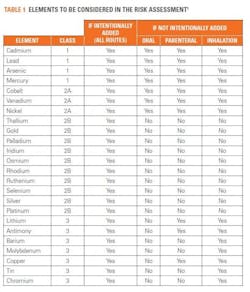
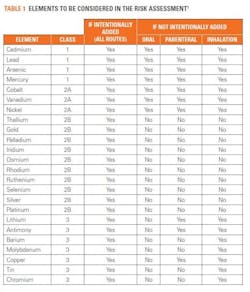
The USP has produced documentation to assist with risk assessment for the 24 elemental impurities that need to be taken into consideration (see table 1). These include the 15 elements listed in USP <232> and nine others, and these have been classified in accordance with their toxicity and the likelihood of their occurrence in the drug product. The USP has also provided risk assessment guidance based on the route of administration of a drug (oral, parenteral or by inhalation). In this assessment, if an element is intentionally added during production, the manufacturer needs to test for it, whereas if a particular element is not added during the drug production process, it may not need to be considered in the risk assessment. However, arsenic, cadmium, lead and mercury must always be tested for because of the risk of their introduction from environmental sources such as water.
Each element poses its own set of potential issues: for example, it is not always necessary to test for gold, however when testing for mercury, gold must be added as it stabilizes the sample. Therefore, when testing for mercury and gold, separate samples must be prepared so that the background levels of gold are not skewed by the stabilizing agent. For several elements, the use of a hydrochloric acid matrix may be beneficial but for others, such as arsenic, the use of hydrochloric acid may lead to an inaccurate test result.

METHOD VALIDATION
The key to validating all test methods is sample preparation and multiple sample preparations may be needed to obtain a validated method for all 24 elements. It is also important to consider whether testing for all 24 elements is required from the start so as not to complicate the overall analysis procedure unnecessarily. In addition, further testing may not be needed if a manufacturer can demonstrate compliance through process monitoring and supply-chain control.
The new USP describes two analytical procedures for the evaluation of elemental impurities: inductively-coupled plasma optical emission spectrometry (ICP-OES) and inductively-coupled plasma mass spectrometry (ICP-MS). The USP states that by using a validation study, an analyst can confirm that the analytical procedure described is suitable for the specified material. Unfortunately, this means that all sample types have to have their own validation study performed. Many methods can be used to test multiple sample types, however each one will need to be validated individually to determine whether the method is the appropriate one to use.
MAKE YOUR MIND UP
It will be up to each manufacturer to determine which approach to adopt when performing elemental impurity analysis. Many excipient and API manufacturers are now beginning to test their materials and will have appropriate information available that they can give to drug manufacturers. However, if the right data are not available, or the manufacturing process may contribute too many elemental impurities to the final drug product, testing that final drug product for impurities may be the better option.
[javascriptSnippet]