Metal Residue: How Much is Too Much?
Contamination of medicinal products with heavy metals may arise from metals deliberately added as catalysts or reagents. Natural occurrence in source materials or processing equipment including vessels, pipes or metal connections to tubes or hoses may be further causes for metal residues. The presence of heavy metals may exert toxicological effects and therefore should be excluded or limited to an acceptable threshold. For many existing substances, approved specifications are provided by the pharmacopoeias in various regions of the world. The following compares current requirements for pharmaceutical substances (APIs, excipients and process chemicals) and describe new guidelines such as those from USP that will take effect in 2014.
Metals in medicinal products or human nutrition can be beneficial or problematic: On one hand, they are used directly as active substances in drug products to exert a beneficial effect, or they are necessary as minerals or trace elements. Many products on the market used as dietary supplements contain trace elements like iron, copper, zinc, selenium, manganese, chromium, molybdenum or other. Many of these metals are essential as parts of enzymes, vitamins or cofactors. Supplementation of minerals or trace elements is needed when dietary intake is deficient and may be beneficial for compensation of deficiencies.
Metals used in drug substances still have importance in modern drug therapy. For example platin compounds (cisplatin, carboplatin) are administered as highly potent anticancer drugs. Aluminum is widely used in antacids; iron is used for treatment or prevention of iron deficiency and anemia; zinc is part of insulin zinc suspensions; cobalt is part of vitamin B12; gold compounds were shown to be efficacious as anti-rheumatoid drugs.
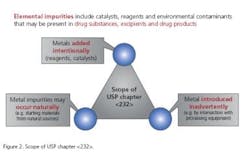
On the other hand, metals in medicinal products may also be present as impurities. Contamination may arise from metals deliberately added as catalysts or reagents. Natural occurrence in source materials (e.g., in minerals or herbals) or processing equipment like vessels, pipes or metal connections to tubes or hoses may be further causes for metal residues. As contaminants, these metals may exert toxicological effects and therefore they should be excluded or limited to an acceptable threshold.
NO THERAPEUTIC BENEFIT
Since there is no therapeutic benefit from metal residues in pharmaceutical products unless administered therapeutically, they should be removed to the extent possible to meet product specifications, good manufacturing practices or other quality-based criteria.
For many existing substances, approved specifications are provided by the pharmacopoeias in each region, such as the European Pharmacopoeia, United States Pharmacopeia and the Japanese Pharmacopoeia. Further, the International Conference on Harmonization (ICH) has published a concept paper (Q3D: Impurities: Guideline for Metal Impurities) on the development of a guideline which would harmonize the limits of metal impurities in the three large economic areas Europe, United States and Japan.
Impurities typically arise from the manufacturing process or from degradation of the substance (Figure 1). An impurity in a drug substance as defined by the guideline ICH Q3A(R2) is any component of the drug substance that is not the chemical entity defined as the drug substance. Quite similar is the definition for an impurity in a drug product. It is any component of the new drug product that is not the drug substance or an excipient in the drug product (ICH Q3B). The impurities which are already controlled in the drug substance need not be monitored or specified in the drug product again, unless they are also degradation products (ICH Q6A).

EMA GUIDELINES
EMA guidelines on specification limits for residues of metal catalysts and reagents were put into place in 2008. The guidelines define maximum acceptable concentrations limits for metal residues arising from the use of metal catalysts or metal reagents in the synthesis of pharmaceutical substances. The objective of the guidelines is to control residues from metals added intentionally.
For products that were already on the market when the new guidelines were enacted, the drug product manufacturer had five years to address. This transition period will end September 1, 2013.
According to the guideline, limits should be provided for metals which are likely to be present due to introduction into the manufacturing process as metal catalyst or metal reagent. What is not in the scope of the EMA guidelines are metals introduced through raw materials, metals arising from interaction with processing equipment, metals added inadvertently or by the environment and packaging materials. The guidelines indicate that these so-called “extraneous metal contaminants” are more appropriately addressed by GMP, GDP or other relevant quality provision.
Thus, only process-related metal residues are in the scope of the guideline to control the sufficient removal of the pharmaceutical substance. Pharmaceutical companies do not need to perform extensive tests on metal residue findings of unknown sources to comply with the guideline. In these cases, companies typically rely on general information from trusted suppliers.
There are four conditions for a metal to be in the scope of this guideline:
1) The metal has to be used in the manufacturing process as catalyst or reagent (regardless of the speciation or form of the element
2) It is likely to be present in the pharmaceutical substance
3) It is not a deliberate component of the pharmaceutical substance
4) It is among the metals of the guideline (see below)
The term “pharmaceutical substances” is defined as a substance that is either an active pharmaceutical ingredient or an excipient. The guideline refers also to metals used in the synthesis of any of the pharmaceutical excipients used during the manufacture of the drug product, but no longer present in the drug product itself and includes 14 metals which are divided in three classes:
Class 1 metals are considered to be metals of significant safety concern. This group includes metals that are known or suspect human carcinogens, or possible causative agents of other significant toxicity. Class 1 is further divided into three subclasses 1A, 1B, and 1C. The subclasses 1A and 1B cover highly toxic or carcinogenic metals. For subclass 1B a group limit is applied, the total amount of listed metals should not exceed the indicated limit.
Class 2 metals are metals of low safety concern. This group includes metals with lower toxic potential to man. They are generally well tolerated up to exposures that are typically encountered with administration of medicinal products. They may be trace metals required for nutritional purposes or they are often present in food stuffs or readily available nutritional supplements.
Class 3 metals represent a minimal safety concern. This group includes metals with no significant toxicity. Their safety profile is well established. They are generally well tolerated up to doses that are well beyond doses typically encountered with the administration of medicinal products. Typically they are ubiquitous in the environment.
For each of these classes, exposure and concentration limits are defined. Limits are based on toxicity of the metal compared to the amount typically ingested, dosage, duration of treatment, and route of administration — whether oral, inhaled or parenteral.
The classification of impurities in three classes was already carried out with the ICH guideline on residual solvents (ICH Q3C). As such, the approach is known with the manufacturers of pharmaceutical starting materials. The classification is solely driven by the toxicological assessments of the specific metals. Quality aspects, for example the color or the possibility to interact on other components like inducing oxidation or catalysis of degradations, is not considered.
EUROPEAN PHARMACOPEIA
In 2009, the European Directorate for the Quality of Medicines and Healthcare (EDQM) established a working party on the topic of heavy metals. Documents were reviewed and finished for adoption by the commission in October 2011. General chapter 5.20 (Metal Residues) implements the text of the EMA guidelines into the European Pharmacopeia; a pharmacopeial text means a higher level of commitment. The general method 2.4.20 on metal catalysts or reagents does not describe a discrete method but offers advice on the choice of method for sample preparation and measurement and defines validation requirements.
It appears that the EDQM may implement monographs based on the EMA guideline fairly soon. The ICH Q3D guideline will replace the EMA guideline in chapter 5.20 in at some point. The conventional heavy metal test will be abolished once the ICH regulations are implemented in the Pharmacopeia.
The EMA guideline seems appropriate to control the risk of elemental impurities in pharmaceutical substances produced by a chemical production process. The list of elements should be extended by metals frequently used in the production of pharmaceutical substances (e.g. lithium, boron, aluminum and silver). The coverage of iron and zinc is questioned since both elements pose a minimal toxicologic risk and Zn is even not very likely to be present. The classical heavy metal test is considered not to be meaningful for control of these elements and should be removed.
An addendum to the general monograph “Substances for Pharmaceutical Use” (2034) is prepared, making the provisions of general chapter 5.20 mandatory. Substances of pharmaceutical use for veterinary use only will be exempted since these substances are out of the scope of the EMEA guideline.
U.S. PHARMACOPEIA
In January 2010, USP proposed new general chapters on elemental impurities limits and methods. The revisions focused on two key areas:
• Update the methodology used to test for elemental impurities in drugs and dietary supplements to include procedures that rely on modern analytical technology
• Setting limits for acceptable levels of metal impurities in drugs and dietary supplements
The monograph on elemental impurities limits ( <232> ) applies to drug products, drug substances and excipients; compliance, however, is required for drug products only (Figure 2). A risk-based approach for testing strategy was adopted.
General chapters <232> and <233> have been published on the USP website in a finalized version since April 2012. A new provision will be added to General Notices that will make these General Chapters applicable to all official articles recognized in USP and/or NF. Because the General Notices provision making these chapters applicable to articles in USP and NF, the date on which users will be required to meet the requirements of these Elemental Impurities chapters will be May 1, 2014.
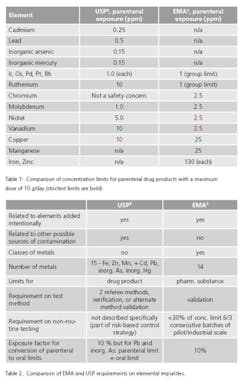
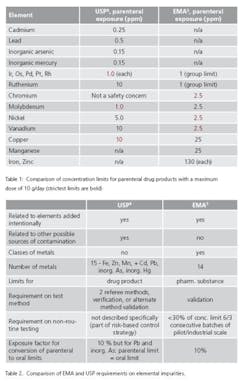
In the final version, there is no longer a classification. The elements included are those of the EMA guideline but without iron, zinc and manganese due to low toxicity. Additional elements are inorganic arsenic, cadmium, inorganic mercury and lead. Table 1 compares the concentration limits for parenteral drug products with a maximum daily dose of 10 g/day are compared.
In addition to the EMA guideline in which testing is required only if these metals are intentionally added during manufacture, the control strategy of the USP chapter <232> also comprises elemental impurities that may occur naturally or are introduced inadvertently (Figure 2).
When elemental impurities are known to be present, have been added, or have the potential for introduction, assurance of compliance to specified levels will be required for the drug product. The conventional heavy metal test will be abolished.
While the limits do not apply to excipients and drug substances but only drug products, it was noted that elemental impurity levels present in drug substances and excipients “must be known and reported.” With regard to these components, limits remain a matter of negotiation between the drug product manufacturer and supplier of pharmaceutical starting materials. Table 2 compares EMA and USP requirements.
ICH GUIDELINES
In 2009, the International Conference on Harmonization issued a plan for the establishment of a new guideline on metal impurities. Current status of the ongoing discussion can be summarized as follows:
• The guideline will cover all drug products including biotech products, but will exempt herbals, radiopharmaceuticals and conventional vaccines
• The guideline will cover PDE and control strategy, but will not cover testing strategy and analytical methods
• The guideline will be applicable to new drug products, but will not be applied to existing products and clinical trials
• The guideline will cover the general risk of contamination with metal impurities
In early 2012, draft guidelines were published for commentary. The guidelines cover drug products and the sources of metals being catalysts and reagents, but also those added inadvertently. PDEs and limits were set for 27 elements, with some limits being stricter and other being less strict. New limits were added for 12 elements and different conversion factors from parenteral to oral limits, depending on the oral bioavailability provided.
OUTLOOK
Regulations governing the presence of heavy metals in drug products are evolving. USP and ICH are setting strict acceptance limits for drug products, and ICH has outlined a risk-based control strategy for up to 27 metals.
The pharmaceutical industry is expected to comply with the new USP requirements from May 2014 on. A risk-based control strategy will be more likely applied than full testing of all elements for each batch. However, a risk-based approach has to be demonstrated to be appropriate. Most considerations at pharmaceutical industry will probably be at excipients. Performing of (inorganic) impurity profiles is currently not required for excipients and not a common practice at many excipients manufacturers. For APIs, the risk of contamination may be less due to the required full validation program of manufacturing process. Impurity profiles for inorganic impurities are more likely to be performed with APIs, predominantly for known impurities from the manufacturing process (among others catalysts, heavy metals, inorganic salts). Actions of excipient manufacturers to set up an appropriate risk-based control strategy shall be supportive to fulfill the requirements addressed to pharmaceutical industry.
As a result of these changes, drug product manufacturers will need proper analytical and regulatory support to achieve compliance for the pharmaceutical substances they use. In parallel, suppliers of raw materials to pharmaceutical manufacturers should be able to state that metals were either not used as catalysts or reagents or are consistently removed. An impurity profile should be available showing evaluation against guideline limits.
ABOUT THE AUTHORS
Ulrich Reichert is a pharmacist and the Head of Regulatory Experts and Customer Audits at Merck Millipore. He has more than 10 years experience in leading analytical laboratories at Merck KGaA where he was responsible for release of pharmaceutical starting materials, GMP and IVD products. He then worked with Regulatory Services responsible for global regulatory projects within Merck Millipore.
Najib Sehat is Director for Regulatory & Technical Services at Merck Millipore. Najib has been with Merck Millipore for over 13 years and has held various leading positions of increasing Regulatory responsibility in various Merck Chemical Divisions. He is a member of various professional organizations such as IPEC Europe, IFT (USA), German Chemical Association, Global Steering Committee of EXCiPACT and member of and deputy Board of Director for the Rx-360 Consortium.
References
1 U. Reichert: Implementing the Guideline on the Specification Limits for Residues of Metal Catalysts or Metal Reagents (EMEA/CHMP/4446/2000, Master Thesis, University of Bonn, June 2009
2 ICH Harmonized Tripartite Guideline. Q3A(R2) – Impurities in New Drug Substances, Step 4 version, 25 October 2006
3 ICH Harmonized Tripartite Guideline. Q3B(R2) – Impurities in New Drug Products, Step 4 version, 02 June 2006
4 ICH Harmonized Tripartite Guideline. Q6A – Specifications: Test Procedures and Acceptance Criteria for New Drug Substances and New Drug Products: Chemical Substances, Step 4 version, 06 October 1999
5 EMEA/CHMP/SWP/4446/2000, Guideline on the specification limits for residues of metal catalysts or metal reagents. Committee for Medicinal Products for Human Use (CHMP), London, 21 February 2008
6 ICH Harmonised Tripartite Guideline. Q3C(R5) - Impurities: Guideline for Residual Solvents, Step 4 version, 4 February 2011
7 The United States Pharmacopeial Convention: Draft <232> and <233>, Pharmacopeial Forum 36(1), 2010
8 The United States Pharmacopeia, Chapter <232> and <233>, 2nd supplement to USP 35-NF 30, Rockville, MD: U.S. Pharmacopeial Convention, Inc.; 2012
9 M. Türck: Elemental impurities in Substances for pharmaceutical use – Current trends in pharmacopoeial testing. Slides to oral presentation, Bern, 24th October 2011.