Leveraging Platform Analytical Methods for Biopharma QbD
Monoclonal antibody (mAb)-based therapeutics are the dominant class of molecule in the biopharmaceutical market today. Year-on-year the number of approved mAb-based therapeutics continues to grow and 2017 is set to be a record year with eight approvals already granted.
It is well documented that mAbs are composed of a large number of variants which are an inherent property of this class of therapeutic products. Variants can arise through post-translational modifications (PTMs) during manufacture and through physical or chemical modifications as a result of the purification, formulation and storage processes. Many of these variant forms have been determined to have an effect on drug safety or efficacy and are termed critical quality attributes (CQAs). The CQAs are monitored throughout development, manufacture and lot release. While each mAb therapeutic is clearly unique in its targeting and activity, the physicochemical properties of mAbs can often be described within relatively narrow ranges.
With a keen emphasis on Quality by Design (QbD), and driven by a focus on patient safety, the regulatory bodies such as the FDA and EMA impose tight rules and regulations around the understanding and monitoring of mAb CQAs. A key aspect of biopharmaceutical QbD, which is yet to be truly leveraged, is the use of so-called “platform” strategies for CQA determination. This article will explore the importance of high-performance liquid chromatography (HPLC) in mAb CQA determination and monitoring, the benefits of implementing well-developed platform mAb HPLC methods and their potential scope and application.
HPLC FOR CQA DETERMINATION
The International Conference on Harmonization of Technical Requirements for Registration of Pharmaceuticals for Human Use (ICH) define a CQA as “a physical, chemical, biological or microbiological property or characteristic that should be within an appropriate limit, range or distribution to ensure the desired product quality”.
HPLC methods represent the most convenient and efficient approach to characterizing many of the key CQAs and are routinely used for charge variant, peptide mapping and aggregate analyses, to name but a few. Today drug manufacturers are challenged with the time it takes to develop and optimize the necessary CQA methods for individual mAbs, or variants of a mAb, for characterization and confident routine (process analytical technologies/lot release) monitoring. Exploiting the similar physicochemical properties of all current therapeutic mAbs, and building platform methods around relevant standards, whereby the hardware, consumables/reagents, software and the underlying methods are all standardized, will provide drug manufacturers with numerous benefits:
- shorter time to market (faster development)
- higher cost predictability for each new biologic entity (NBE)
- the ability to standardize operations and staff training
- reduced disruption to current operations
- less wastes
- the flexibility to set up and test complete analytics platforms before they are commercially deployed or outsourced
Aligned with the FDA’s push for QbD, HPLC and mass spectrometry, instrument vendors are now working with industry partners to develop platform methods for major CQA workflows.
MAKING CHARGE VARIANT ANALYSES BASIC
Charge variant analysis (CVA) with a salt gradient elution, although widely used, was never regarded as a platform method that could be used with any mAb product. The different isoelectric points for the proteins required careful and lengthy method optimization for each mAb. Introduction of pH gradient elutions for CVA has changed this perception and permits a single method to be used as a global starting method for any mAb product. A pH gradient can be set up such that it covers a pH range wherein, at some point, any target mAb and its associated charged variants will reach their isoelectric points, become uncharged and so elute from the column. The technique is essentially one of isoelectric focusing and is also a powerful variant concentration technique. Further optimization for individual mAbs can easily be performed from an initial scouting gradient. These characteristics have firmly placed CVA into the list of routine methods with potential platform applicability. This has recently progressed further with the availability of commercially available buffer cocktails, which offer exceptional linear control over a pH gradient.
SIMPLIFYING MAPS
Peptide mapping is a workflow used for all protein therapeutics which can measure several CQAs necessary for complete characterization. The analysis can be implemented in a HPLC - ultraviolet (UV)-only method once the peaks have been identified by mass spectrometry, which is the preferred route in quality control laboratories.

The level of reproducibility shown in Figure 2 for five different analysts performing a manual protein digest was previously unheard of. This has become possible due to advances in UHPLC hardware and, more recently automation of protein digestion. This removes the inherent complexity in peptide mapping and firmly positions the technique as a robust, easy-to-use QC methodology.
Recently there has been a surge of interest in a multi-attribute method (MAM), in which the addition of high resolution accurate mass (HRAM) mass spectrometry information in a peptide mapping approach is used to gain more information from a single injection. Filings to begin clinical trials using this approach have now been placed with the FDA.
Aggregation with Stamina
With the advent of single-injection MAMs such as peptide mapping, it is easy to imagine a QC lab without the need for numerous methods during manufacture and lot release, as is common place today. However, one CQA that is very difficult to evaluate by such methods — and one that has a serious implication for patient safety — is the aggregation profile of the drug product. Therapeutic protein aggregates are degradation products that arise from partial unfolding and/or additional conformational changes in protein structure, exposing hydrophilic groups and facilitating the formation of non-covalent protein-protein bonds resulting in dimers, trimers and further high order structures. This degradation can occur due to sub-optimal conditions at many stages throughout the manufacturing process, and it is therefore critical to optimize at each stage including: clonal selection, upstream and downstream processing, formulation as well as transport and storage to ensure the lowest possible levels of aggregation in the final drug product. It is expected that a higher level of aggregation can reduce product efficiency by lowering the effective concentration of the product. Elevated aggregation has also been found to trigger immunogenic response in some patients. Thus, it is one of the CQAs that must be monitored and reported during each lot release, in order to comply with regulatory requirements.
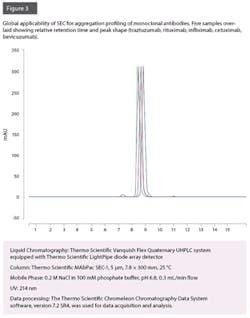
However, due to high costs associated with drug development and production and the pressure to reduce the costs to compete with biosimilars, it is paramount that the hardware and consumables required to perform this analysis are able to offer a robust platform method that can run continuously for many weeks, with zero unplanned down-time and without the need to change consumables or wear parts. Column fouling has been commonplace in SEC analysis, and suppliers of SEC columns have reported column stability lifetimes of approximately 550 injections (without a column guard) and up to 902 injections (with column guard). Additionally, a bio-compatible system is required to withstand high salt concentrations of the eluent, without the degradation of wear parts.
Recently it has been shown that with use of the latest instrument and column technologies, it is possible to run almost 2,000 injections before observed reduction in column performance without the need for a guard column.
FUTURE OF PLATFORM METHODS
The implementation of well-developed platform mAb HPLC methods, addressing the need to monitor various CQAs, has been described. Recent advances in instrumentation and techniques for CQA characterization are significantly increasing the applicability of platform HPLC methods, e.g., CVA and peptide mapping.
Platform flexibility, with the ability to seamlessly incorporate user-friendly HRAM mass spectrometry, provides additional benefits, enabling generation of information on multiple CQAs per single injection.
Despite the ability to incorporate platform methods to simultaneously address various CQAs, certain CQAs maintain the need for dedicated assays. Instances such as aggregation profile assessments, with their extremely high throughput demands, require extremely robust platform methods, which are now available.
REFERENCES
- ICH, ICH Harmonized Tripartite Guideline Q8: Pharmaceutical Development, Step 4 version (August 2009).
- Farnan, D., and Moreno, G. T (2009) Multiproduct high-resolution monoclonal antibody charge variant separations by pH gradient ion-exchange chromatography. Anal Chem; 81(21):8846-57.
- Millán, S., Farrell, A. and Bones, J. (2017) Using the NISTmAb reference standard to demonstrate a simple approach to charge variant analysis. Thermo Scientific Application Note 21684 (https://tools.thermofisher.com/content/sfs/brochures/AN-21684-LC-NISTmAb-Charge-Variant-Analysis-AN21684-EN.pdf).
- Rogers, R.S., Nightlinger, N.S., Livingston, B., Campbell, P., Bailey, R. and Balland, A. (2015) Development of a quantitative mass spectrometry multi-attribute method for characterization, quality control testing and disposition of biologics. MAbs; 7(5):881-90.
- Farrell, A., Bones, J., Cook, K., Patel, S., Schwahn, A. and Bardsley, J. (2017) High-precision, automated peptide mapping of proteins. Thermo Scientific Application Note 21682 (https://tools.thermofisher.com/content/sfs/brochures/AN-21682-SP-LC-MS-Peptide-Mapping-Proteins-AN21682-EN.pdf).
- Blank, M. (2017) Advanced QA/QC characterization MS in QC : Multi Attribute Method. Thermo Scientific Global BioPharma Summit Presentation (https://assets.thermofisher.com/TFS-Assets/CMD/Reference-Materials/PP-72521-MS-QA-QC-Multi-Attribute-Method-BioPharma2017-PP72521-EN.pdf).
- Arnaud, C. H. (2016) Mass spec weighs in on protein therapeutics. Chemical & Engineering News; 94(22):30-34 (https://cen.acs.org/articles/94/i22/Mass-spec-weighs-protein-therapeutics.html?h=418213537).
- Farrell, A., Bones, J., and Cook, K. (2017) The importance of correct UHPLC instrument setup for protein aggregate analysis by size-exclusion chromatography. Thermo Scientific Application Note 21602 (https://tools.thermofisher.com/content/sfs/brochures/AN-21602-LC-SEC-mAbs-Instrument-Setup-AN21602-EN.pdf).
- Farrell, A., Bones, J., and Cook, K. (2017) A universal chromatography method for aggregate analysis of monoclonal antibodies. Thermo Scientific Application Note 21601 (https://tools.thermofisher.com/content/sfs/brochures/AN-21601-LC-SEC-mAbs-AN21601-EN.pdf).
- Improving the Lifetime of UPLC Size-Exclusion Chromatography Columns using Short Guard Columns (2011) Waters Technology Brief (http://www.waters.com/webassets/cms/library/docs/720004034en.pdf).
- Farrell, A., Jakes, C., Ley, A., De Pra, M., Steiner, F. and Bones, J. (2017) Lifetime stability of size exclusion chromatography columns for protein aggregate analysis. Thermo Scientific Application Note 72362 (https://assets.thermofisher.com/TFS-Assets/CMD/Application-Notes/AN-72362-LC-Protein-Aggregate-Analysis-AN72362-EN.pdf).