DOE Reduces Bioequivalent Generic Development Time to 4 Months
A generic pharmaceutical manufacturer recently hired VerGo Pharma Research Laboratories Pvt. Ltd to develop a bioequivalent with different polymorphic forms for an anti-depressant drug that had previously been patented in crystalline form only. Bioequivalence requires that a drug be pharmaceutically equivalent and that it be delivered at the same rate and same level of bioavailability so that its efficacy and safety can be expected to be the same as the original product. Using conventional one-factor-at-a-time testing methods, it would have taken several years to determine the right combination of inactive ingredients to achieve the required in-vitro dissolution and in-vivo plasma drug profile. By using design of experiments (DOE) to reduce the number of tests required (to determine the effects of inactive ingredients on bioavailability in both fed and fasting conditions), VerGo was able to cut this development process to only four months.
FORMULATION DEVELOPMENT CHALLENGE
In this formulation development application, a number of generic drug producers had worked with other contract research companies who had been able to achieve the right concentration of the drug in the blood of volunteer patients in the fed condition but were not able to achieve the right concentration in the fasting condition. VerGo staff speculated that the reason the other contract researchers were not able to find the right formulation is that those firms were testing the effects of one ingredient at a time and thus were missing potential effects caused by interactions between ingredients.
VerGo dramatically reduced the time required for formulation development by using DOE to quickly identify interactions between ingredients and reduce the number of experiments required to optimize the formulation. Stat-Ease Design-Expert software was used to design an optimal experiment with three factors, each consisting of the concentration of an inactive ingredient, an insoluble diluent, a mild disintegrating agent and a super-disintegrating agent.
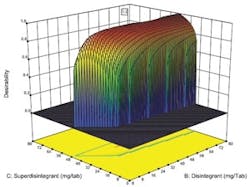
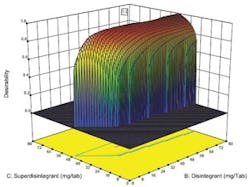
Desirability function plotted against concentration of two key ingredients.
Optimal designs build test plans based on a statistical criterion and the model chosen by the experimenter. Design-Expert software was selected because it offers a wide range of experimental designs and because it makes it easier for users without a statistical background to design an experiment and analyze the results.The software selected values for a total of 20 runs with the diluent ranging from 0 to 194 milligrams per tablets and the two disintegrating agents ranging from 0 to 80 mg per tablet. The experiment included 5 replicates, which were used to measure the reproducibility of the results.
VerGo scientists executed the batches (see Table) sequentially based on the run number to minimize the experimental errors. Under my observation, they took the above-mentioned compositions provided by the software and produced the tablets for each experiment while keeping the manufacturing process parameters constant. The dissolution of each tablet was measured in the lab at 1.6 pH at 20, 45 and 90-minute levels and at 5.5 pH at 20 and 45 minutes (see Figure).
IDENTIFYING THE OPTIMAL FORMULATION
After running the experiments, we entered the results into the software along with the ideal values for the dissolution rate at each pH value/time point pair. These dissolution rate values were selected to match the values achieved by the original drug based on the assumption that if the proposed generic performs the same as the original drug in the lab, it is likely to also perform the same in clinical testing. The software generated a prediction of the concentration of each variable required to meet all of the target dissolution values. A new batch of tablets was then produced with the recommended concentration values. These tablets matched the desired dissolution profile within +/- 5%, which is within the acceptable margin of error.
VerGo decided to test the proposed pharmaceutical in the presence of two surfactants that are present in the human body with the goal of ensuring its performance in clinical testing. We did not use these surfactants in the original designed experiment because they were too costly and unstable to run a complete experiment. We found that the surfactants speeded up the dissolution profiles of the test formulation as compared to the reference formulation.
Hence, we reduced the value of the super-disintegrant in order to reduce the dissolution profile back to the target value. The sensitivity of the formulation to the super-disintegrant as determined by the designed experiment was used which, in fact, reduced the surfactant by the amount suggested by the software and had the desired effect of hitting the target dissolution profile within +/- 5% with the surfactants present.
A larger batch of tablets was then prepared with this formulation for clinical testing with volunteer patients. The patients took the drugs in both fed and fasting conditions and the concentration of the drug in their blood was measured at set intervals. The results showed that VerGo was the first company able to match the blood concentration levels of the active ingredient to the original pharmaceutical over its full-time profile.
While the formulation study achieved all of its goals, the product has not yet reached the market due to unrelated business and legal issues. However, the success of the project has demonstrated more clearly the advantages of DOE in formulation development. This project was successfully completed in four months compared to two years for similar projects (undertaken without the benefit of DOE). VerGo has seen its formulation development consulting contracts increase as clients request that drug products be developed using DOE strategies. In addition, the use of DOE at VerGo has spread beyond formulation development; for example, the technique has been used successfully several times in developing new analytical methods.