Editor's Note: See the end of this article for data from our recent RMM survey, a list of vendors and their rapid method technologies, and links to related resources.
The past few years have seen a wide array of rapid microbial methods (RMM) reach the market. Designed to reduce the time required for traditional microbiological testing, these newer methods (Box, below) are based on widely different technologies but they can reduce plant downtime significantly, and cut testing time from weeks to days or hours.
At this point, more pharmaceutical manufacturers are using RMM’s, as demonstrated by a growing number of new drug approvals (NDA’s) specifying use of the technology, but there has been no mass stampede to convert existing operations.
Concerns about cost, validation and acceptance by global regulatory agencies are all tempering enthusiasm.
Apart from one method that was commercialized last year by Battelle Laboratories (Box, below), there is no single RMM that can detect and count microbes. In many cases, testing is considered destructive because samples cannot be stored or analyzed after testing.
Despite FDA’s affirmed support of use of RMM, and numerous guidance documents and requirements from agencies around the world, there are lingering concerns about European regulatory approval.
|
Spectroscopy, REBS and the Optical spectroscopy is becoming a more viable technique for rapid microbial identification and monitoring. Infrared spectroscopy is being studied for analysis of bulk materials, but Raman is being applied to detect individual cells. Last year, researchers at the Electronics, Sensors and Information Systems division of Battelle Memorial Laboratories commercialized the REBS, the Rapid Enumerated Bioidentification System, based on Raman spectroscopy. The technology is nondestructive, and detects, counts and identifies microbial particles larger than 300 nm in diameter. The system takes about five minutes to analyze each particle, and less than $10 per sample. A major advantage is that it allows for sample archiving and secondary confirmation of results. Each device is modular and weighs less than 100 lbs. |
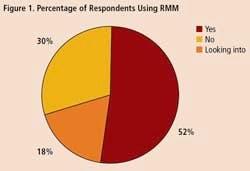
To get some idea of the impact that RMM’s are having within the drug industry, Pharmaceutical Manufacturing recently surveyed readers on their use of rapid methods. Professionals at 34 drug companies responded (results are summarized on below). Only 18% of respondents say they are not interested in the technology. Over half of those who responded to the survey say they are already using RMM, and another 30% are evaluating these technologies.
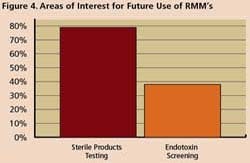
Survey respondents say they are using RMM mostly for non-sterile process applications such as WFI, for sterile product testing and environmental monitoring. However, 77% said they are interested in applying RMM for sterility testing, while 36% are exploring use of RMM in endotoxin screening.
To put survey results in perspective, we asked two PDA Committee experts on RMM for their comments and views: Jeanne Moldenhauer, VP of Excellent Pharma Consultants (Mundelein, Ill.), who wrote a chapter dedicated to RMM’s in “Priciples of Bacterial Detection,” published in September 2008 by Springer, and and Michael J. Miller, Ph.D., President of Microbiology Consultants, LLC (http://microbiologyconsultants.com; Tampa, Fl.), and the editor of the “Encyclopedia of Rapid Microbiological Methods,” published by DHI Publishing in 2006.
PhM: Pharma is notoriously conservative about applying new technologies. Is there a mindset that is preventing more manufacturers from using RMM, and is perceived cost an issue?
JM: Cost is definitely an issue. Most companies use ROI to approve new investments, and its not always easy to prove that RMM’s will pay for themselves within a few years, particularly for sterility testing. One can expect to pay between $150,000 and $400,000 for the typical system alone. Validating the systems will cost about the same.
Accountants don’t typically assign a dollar value to controlling risk, so quantifying the benefits of what may be a good investment can be difficult. Generally, it is done based on projections of reduced inventory, hold time or headcount.
MM: You have to weigh costs for many things—automation, higher throughput, and elimination of headcount.
PhM: Many of our survey respondents expressed concerns about validating RMM’s. Aren’t there many guidance documents already out there that support their use?
JM: Several guidance documents are available. Yet, there are some points that may remain unclear. For example, compendial requirements dictate that statistically-based tests be used to validate RMM’s. Basically, the industry took chemistry-based tests and applied them to microbiology, and the methods can be difficult to use when working with low counts. In sterility tests, for example, you’re dealing with 0 or 1 levels. Statistics need to be “debugged,” since there was no general acceptance at the time the requirements were published.
MM: These issues are being addressed by the PDA Committee on Technical Report No. 33. The document came out in 2000, but is being revised to clarify validation and to provide more guidance on use of statistics. Revisions are expected by the end of this year, or early next.
The USP Chapter <1223> and European Pharmacopoeia Chapter 5.1.6, which are similar, also provide guidance on how to qualify rapid methods. Another resource is FDA’s guidance on aseptic processing, which contains language that specifically mentions “methods such as RMM”.
PhM: How can vendors help validate RMM’s?
JM: It can be challenging, especially for sterility testing, to prove that RMM’s can detect low counts in, for example, an air sample because most pharmaceutical company labs can't develop a challenge that low.
MM: Some vendors are now using standardized cultures for testing. Last year, BioVigilant Systems, Inc. conducted USP <1223> validation studies using low levels of standardized cultures. Results were submitted to the FDA under a Drug Master File (DMF). Any new user of the system can now refer to the DMF so that they do not have to repeat the standardized testing. However, end users will have to conduct their own testing in tandem with the methods they currently use.
PhM: If FDA has openly supported RMM’s, how are other global regulatory agencies approaching the technologies?
JM: Several recent conferences have discussed the issue of European approval. There have been approvals on the books for quite a few years, and there’s wide acceptance of RMM for bioburden monitoring, but in some cases, it can be hard to get RMM approved in Europe. Part of the problem may be that, unlike FDA, European agencies don’t have a comparability protocol in place, which can make it more difficult for manufacturers to make broad spectrum changes.
MM: It can be confusing in Europe, but some regulators there have made it clear that if one wants to move from traditional methods to RMM’s for microbiological tests that have not been included in a marketing authorization, that change can be managed within internal corporate control processes.
PhM: Is real-time release the ultimate goal for RMM? Will these systems allow companies to apply PAT to sterility testing and aseptic manufacturing?
JM: Some RMM’s used for endotoxin testing provide near real-time, online measurement capabilities. Raman spectroscopy and other optical methods can also be used.
MM: The biggest bang for the buck is not going to be sterility testing, since it is not a rate-limiting step [for many companies] for getting product out the door. The rapid methods available today are perfect for alternatives to conventional in-process testing—for example, environmental monitoring, water testing, or bioburden testing.
Similar to the adoption of PAT, we are moving away from end-release testing to in-process testing. End-product sterility testing is not a good predictor of your overall control of your processes.
The ultimate goal is to be able to use rapid methods in the in-process phase, so that you will know during manufacturing or filling that you don’t have microorganisms in the process. If enough data can be generated, then, at some point in the future, the industry will not need end-product data for sterility [confirmation]. That’s why we’re moving toward the concept of parametric release. Some companies are already doing this for non-sterile products, and I see this happening next in sterile products.
In ten years we’ll have used enough in-process, real-time methods and many of these enough methods will have been accepted by regulatory agencies. When you add all of these individual RMM pieces together, the results will tell you whether or not your process is under control from a microbiological standpoint. You’ll be able to justify the elimination of no end-product testing.
If [in 10 years] pharmaceutical manufacturers are not in a position to challenge the perception that we do not have enough data to eliminate end-product microbiology testing, then we’re not doing our job.
PhM: Wouldn’t parametric release require the use of technologies that provide quantitative as well as qualitative data?
MM: The Holy Grail would be a system that could handle identification, qualitative and quantitative testing. We’ll get there, but it’s going to take existing technology companies to talk to each other to see how to merge detection, enumeration and identification platforms.
PhM: What’s the likely time frame for that?
MM: I’d give it 10 years. It’s possible to take two systems [quantitative and identification] and use them in tandem. I’ve actually made introductions of one company to another, and have tried to explain to them that if they work together, the sum may be much better than the parts
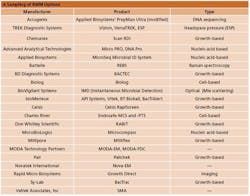
A Sampling of RMM Options
Pharma Not Yet Sold on Rapid Methods
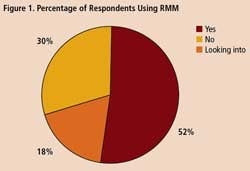
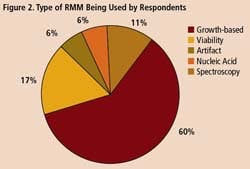
A survey of 34 pharmaceutical manufacturing professionals found that most were already using RMMs (Figure 1), mainly growth-based, viability and spectroscopic types (Figure 2), and using them mainly for environmental monitoring and non-sterile testing (Figure 3).
Respondents agreed that the most serious concerns with RMM centered on validation and regulatory agency response, both of which topped the list for 77% of respondents. System compatibility was noted by another 64% as a top issue.
Cost was also noted as a significant issue. Although one respondent said that ROI was more important than cost, others noted poor exchange rates and the challenges of proving cost vs. benefits.
“The technology is expensive, unless you have sufficient volume to justify its capital and operational costs,” wrote one respondent. “I would consider using a local contract test lab instead.”


Additional Reading
Miller, M., ”RMM for a New Generation,” Pharmaceutical Manufacturing, February 2006.
http://www.pharmamanufacturing.com/articles/2006/019.html
Miller, M., Encyclopedia of Rapid Microbiological Methods. PDA and Davis Healthcare International Publishing. 2005.
The Microbiology Network
http://microbiol.org
The Microbiology Forum
http://microbiol.org/PMFList_info.htm