A Framework for Technology Transfer to Satisfy the Requirements of the New Process Validation Guidance: Part 1
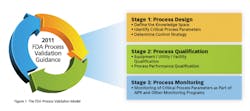
The technology transfer of a process, whether it is from R&D to commercial manufacturing or to another site or contract manufacturing organization (CMO) is a critical step in the lifecycle of any drug product, involving many steps. As major blockbuster drugs come off patent and large pharmaceutical companies look to bolster their pipeline through acquisition, the control and consistency of development data can vary dramatically. To make matters more complicated, the new Process Validation (PV) Guidance issued by FDA in January 2011 now defines three major stages of process validation that must be satisfied to consider the process validated.
With the present article, we will lay out a practical approach that addresses this complexity and propose to discuss and summarize the diverse factors required to describe the process, establish the control strategy and specify the acceptance criteria to successfully transfer a legacy or newly acquired process to another process train and satisfy the new guidance.
To illustrate, we will take a closer look at the methodologies employed and the challenges encountered as part of a recent technology transfer process validation exercise executed for a legacy product for a client organization, with references to the business unit and technology transfer team assembled for the project.
Through this real life example, Part I will discuss the approach taken to establish the design and control space for the final process. Part II will describe the Process Performance Qualification (PPQ) study design and acceptance criteria for Stage 2 and the approach taken to satisfy Stage 3 of the new PV guidance.
The New PV Model
Under the 1987 guidance PV could be characterized as “quality by sampling and testing” while the new guidance would more appropriately describe validation as “quality by design and control.” Let’s look closer at the three distinct stages that make up the new definition of process validation:
- Stage 1 Process Design: The commercial manufacturing process is based on knowledge gained through development and scale-up activities
- Stage 2 Process Qualification: The process design is evaluated to determine if the process is capable of reproducible commercial manufacturing
- Stage 3 Continued Process Verification: Ongoing assurance is gained during routine production that the process remains in a state of control
- Quality, safety, and efficacy are designed or built into the product.
- Quality cannot be adequately assured merely by in-process and final product inspection and testing.
- Each step of a manufacturing process is controlled to assure the finished product meets all quality attributes including specifications.1
“Manufacturers of legacy products can take advantage of the knowledge gained from the original process development and qualification work as well as manufacturing experience to continually improve their processes. Implementation of the recommendations in this guidance for legacy products and processes would likely begin with the activities described in Stage 3.1”
The big difference with legacy products vs. NMEs as they relate to PV is that the baseline data gathering activity begins in Stage 3 of the PV lifecycle rather than Stage 1.
The Technology Transfer Framework
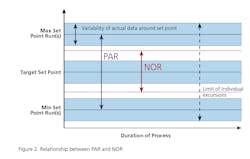
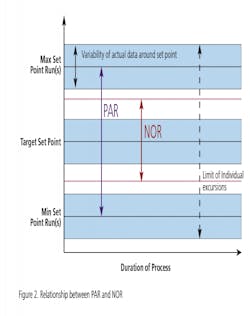
Gone are the days of simply comparing product performance against its release specification. The objective of technology transfer is to acquire the necessary process and product knowledge to establish a PAR and NOR for each unit operation that is consistent with the predicate process being transferred. Thus the new PV guidance requires the demonstration of process reproducibility in the PPQ phase of Stage 2. Reproducibility effectively requires establishing acceptance criteria that are consistent with the process stability demonstrated in the predicate process. Reproducibility must be defined for within lot and between lot variability as part of the PPQ exercise. The technology transfer framework used for this project is based upon Pharmatech Associates’ PV model shown below in Figure 3 and will be discussed as follows: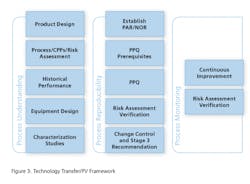
- Greater than 50 percent Active Pharmaceutical Ingredient (API)
- Round 200 mg tablet
- Coated to mask taste
- 12-hour drug release with the following specifications:
- 4 hour dissolution 20-40 percent
- 8 hour dissolution 65-85 percent
Technology Transfer Model: Process Understanding
Product DesignThe technology transfer package included the formulation, raw material, API and finished product specifications and master batch records. No development report was ever written for the product. The team looked at the Chemistry, Manufacturing and Control (CMC) section of the non-disclosure agreement to understand the composition and functionality of each component of the formulation. The formulation is shown below in Table 1.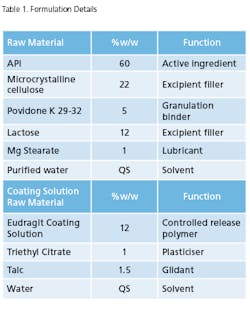
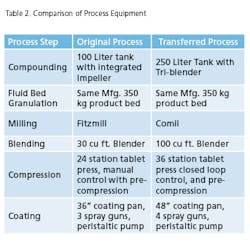

Historical Data Analysis
The absence of development data establishing the PAR and NOR for the CPP can be ascertained to some extent by evaluating the historical behavior of each parameter along with the corresponding behavior of the CQAs for the unit operation. Data should be extracted from multiple batch records to determine whether the process is stable within lot and between lots. In some cases, only mean data or composite data may be available. To do this the team went back into the batch records of approximately 30 lots across a period of one year to extract the necessary data. This exercise also gave some indication as to whether the parameter was truly a CPP, based upon whether it had an impact on the corresponding CQA for the unit operation. The data for each unit operation were plotted as control charts and the process capability was determined. Excursions outside the 3 sigma limit of the control charts were investigated to determine if there were deviations associated with the events. An example of the control chart and capability histogram for fluid bed product bed temperature is shown below in Figures 4 and 5. Capability limits are based on a previously established PAR for the product bed temperature.
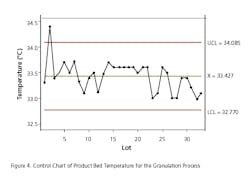
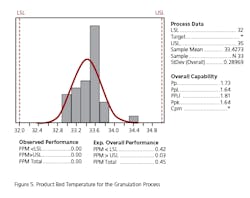
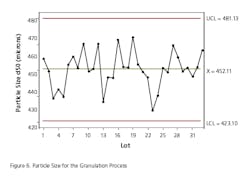
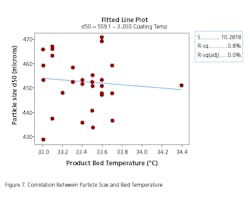
This approach was repeated based upon the parameters that had a medium or high rating in the risk table. For these scale independent parameters the existing PAR ranges were used for the next phase of scale-up studies.
Characterization Studies
For those parameters that were scale dependent additional characterization studies were required to establish PAR and NOR that were consistent with the predicate process. For simply scalable processes like blending, single time-based blend uniformity studies may be adequate to identify the PAR and NOR for the new scale. For more complex unit operations, such as the coating operation, a Design of Experiments (DOE) approach may be more appropriate. The team developed a series of balanced orthogonal experiments to establish the PAR for these parameters. This raises another good point to consider when confirming CPPs. By conducting the historical analysis first it is possible to reduce the number of variables in the experimental design which reduces the number of runs required.
Conclusion
The new guidance is moving the industry toward a quality-by-design philosophy for process validation. This translates to a more parametric approach rather than an attribute-based approach to process design. The application of a risk based model, considering the process and product design at the outset of the technology transfer project allows the application of scientific understanding to filter the potential list of parameters that may affect the process and product CQAs to a limited few. The analysis of historical performance reduces the number of factors that may need to be characterized at the next scale. It also provides a foundation for establishing a baseline PAR and NOR for scale independent parameters when moving to the next scale, factoring in the larger scale equipment design and configuration. Finally, applying a DOE approach to the few remaining scale dependent parameters will establish the corresponding PAR and NOR for the transferred process before moving to the process Control Stage of the roadmap.
In Part II of this case study we will discuss the considerations in developing an effective sampling plan and acceptance criteria for the Stage 2 PPQ along with how to transition to the Continuous Monitoring stage of the new PV guidance.
References:
1. Guidance for Industry: Process Validation General Principles and Practices - FDA, January 2011.
2. Wheeler, Donald, Understanding Variation: The Key to Managing Chaos, ISBN 0-945320-35-3, SPC Press, Knoxville, TN.
3. Chatterjee, Wong and Rafa, Using Operational Excellence to Meet the New Process Validation Guidance, Pharmaceutical Engineering, Sept 2011.