Amidst biologics patent expirations and the push for personalized (yet low-cost) medicines, the age of biosimilars is upon us. The European Union has paved the way with its biosimilar approval pathway, while FDA passed the Biologics Price Competition and Innovation Act (BPCIA) in 2010. The Act codified into law the 351(k) abbreviated regulatory pathway for biosimilars approval, and formally opened the door for biosimilars product approval in the U.S. However, the law provided no advice regarding what requirements the FDA would need for approval, so in early 2012, the Agency issued draft guidances describing CMC considerations in demonstrating biosimilarity to a reference protein product (U.S.-approved or foreign innovator biologic) [1].
Like their reference products, biosimilars are complex, difficult to characterize, typically have more than one biological effect, and frequently generate immune responses. Because of this, the guidance necessarily lacks a list of specific steps for developing biosimilars, leaving developers to integrate quality attributes on a case-by-case basis using the totality of evidence approach. Although approval of a biosimilar will rely on current data of the reference product, the guidance paves the way for producing biosimilar proteins through the use of alternative expression systems and novel manufacturing technologies. To do this, however, developers must ensure they use the principles of Integrated Drug Development to incorporate robust quality considerations in their development programs.This article provides a review of the essentials of developing and manufacturing biosimilars today. We review current animal-, yeast-, and plant-based expression systems, predominant manufacturing technologies, and key quality considerations for developers and manufacturers of biosimilars—all with an eye toward the integration of these elements.Alternate Expression SystemsFor more than two decades, most biologics have been manufactured in well-known, well-characterized, FDA-approved cell lines that were developed by the innovator manufacturers. A review of global product approvals from January 2006 to June 2010 shows the distribution of these cell lines [2].
Animal- or Yeast-Based Alternative Expression Systems
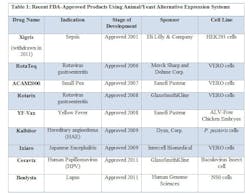
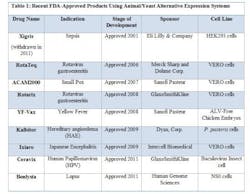
FDA has approved novel products using animal or yeast-based alternative expression systems.
- YF-Vax was developed in Avian Leukosis Virus (ALV)-free chicken embryos. Chicken embryos have historically been used for protein expression and experimentation, and are economical and easily accessible. Most importantly, chicken embryos lack an immune system, making them ideal candidates for production.
- Due to their susceptibility to a wide range of viruses, VERO cells, a kidney epithelial line isolated from the African Green Monkey, have served as a popular expression system for vaccines, such as RotaTeq, Rotarix, ACAM2000, and Ixiaro.
- HEK293 cells (Human Embryonic Kidney cells) have also been used in the manufacture of an approved drug, Xigris. (Note: Xigris was withdrawn from the market in 2011 due to lack of efficacy, not due to any manufacturing deficiencies.) Similar to CHO cells, HEK293 cell lines are easy to culture, have higher rates of expression for proteins of interest, and are easily scaleable.
- The baculovirus insect cell line, used to produce Ceravix for the treatment of HPV, produces large quantities of proteins in cultured insect cells or insect larvae and these proteins are easily purified with tags or using affinity chromatography techniques.
- In 2009, Kalbitor produced from P. pastoris (a yeast) was also approved. These yeast cells can grow to high densities compared to the more common yeast S. cerevisiae. S. cerevisae is also known to release ethanol during the fermentation process, which can deter cell growth and protein production.
- In 2011, Benlysta was approved for the treatment of lupus and was expressed in the mouse myeloma cell line NS0. The NS0 cell line cannot produce endogenous antibodies, making it an attractive platform for protein production. However, as seen with CHO cells, NS0 cells have the potential to generate glycosylated proteins, which are known to produce immunogenic effects.

Although not discussed here, companies are pursuing expression systems outside even these novel systems, including ciliates, alternative yeast and bacterial species, and cell-free expression systems. While in the discovery or preclinical stage, one day these expression systems may have the same degree of success as animal and plant-based systems.
Selected Manufacturing Technologies
Several innovative manufacturing technologies are available for both high throughput screening of recombinant protein variants as well as rapid protein production. Principles of synthetic biology and industrial engineering are used to enhance product expression and development through a controlled, scaleable, and efficient process.
High Throughput Screening
These principles form the basis of Intrexon Corporation’s proprietary UltraVector Platform which offers a dynamic library of modular components to customize, test, and optimize various protein candidates in a cell line of interest based on host cell performance. Combined with its Laser-Enabled Analysis and Processing (LEAP), developers have the option to rapidly identify and select high-secreting, genetically modified mammalian cell lines producing the protein of interest. These techniques are already in use by companies like Adeona Pharmaceuticals, which is leveraging Intrexon’s advanced transgene engineering platform for continuous in vivo cellular production of an enzyme for the treatment of pulmonary arterial hypertension.
Production Technology
The need to get to the market quickly will lead companies to explore flexible, cost-effective manufacturing tools such as single-use technology (SUT). Conventional systems like large-scale stainless steel bioreactors require design and validation and clean-in-place/steam-in-place procedures, whereas SUTs have reduced cleaning validation requirements between product changeovers and batch-to-batch processing. Single-use portable bioreactors, for example, are available at various capacities, providing linear scaleability throughout the manufacturing process for biosimilars using alternate cell lines. cGMP facility operations and infrastructure requirements such as classified clean rooms, dedicated HVAC systems, and gowning are also reduced with SUTs.
According to technology provider Xcellerex, the SUT platform allows production lines to start up within 15-18 months (compared with 3-5 years for conventional systems) and manufacturers benefit from a decrease in total capital cost by 50-75% and operating cost reduction by 20%. This presents biosimilars developers with the ability to accelerate upstream manufacturing processes and dedicate the upfront cost savings towards commercial production needs.
Purification Processing
While high-producing alternative cell lines and less complex upstream manufacturing techniques offer some advantages, downstream purification processing can be a significant bottleneck to CMC development. For some development programs, downstream purification accounts for 80% of the total manufacturing cost [2]. Streamlining purification and reducing the number of steps required in the purification process are two commonly used techniques.
However, more emphasis is being placed on optimizing purification systems. For example, 3M Purification’s Zeta Plus line of single-use, depth-filtration systems can remove cells and selected contaminants and host cell proteins from cell culture media at the primary recovery step using charge-modified depth filters. This system can also be applied to mammalian cell harvest or bacteria, yeast, and insect cell lysates clarification. The specialty media has been shown to reduce host cell proteins, protein aggregates, and endotoxins. This not only provides time and cost efficiency but reduces protein yield loss observed during multiple purification steps.
Quality Considerations for Biosimilar Development
If using a different expression system, it is impossible to generate a biosimilar which is identical to the reference product. However, it is possible to develop highly similar products with no clinically meaningful differences by remembering that “the process is the product.” Upstream production and downstream processing techniques can yield protein fluctuations and impurity profiles that vary among expression systems. When using alternate cell lines, the FDA expects developers to provide sufficient justification that the construct encodes the same primary amino acid sequence as the reference product, with data supporting that minor modifications do not affect safety, purity, or potency of the product. Comprehensive cell line development and characterization data shared early in the development process with the FDA will enable the developers to collect sufficient safety and efficacy data.
Products that are highly similar to reference products can be developed by implementing consistent and complete product characterization testing per International Conference on Harmonization (ICH) quality guidelines. This includes identity testing and structure confirmation during all stages of protein folding (primary through quaternary). A purity assessment accounting for all host cell-related protein availability, and those impurities associated with the protein itself, such as truncated forms, aggregates, or modifications (e.g., glycosylation) should be completed. These impurities could have an effect on protein quality, potency, and the amount of safety and efficacy data required during development. Protein function should also be assessed.
Table 3 lists recommended assays for product characterization, which will vary based on the type of protein and production process.
Once fully characterized, a master cell bank (or seed bank when working with plant expression systems), is generated based on the quality traits desired. This banking system is designed to ensure consistent production of a highly similar protein product. A stability protocol using many of the assays used to characterize the original protein should be in place to control for changes during storage and shelf-life of the product. Agency reviewers expect that the stability protocol assesses phenotypic traits, such as protein production and titre, as well as genotypic stability. DNA sequencing and segregation analysis can be used to monitor potential changes to the protein construct on a DNA level, prior to the expression of the protein itself.
A developer must keep in mind that although a full protein characterization may initially support a highly similar protein product, the expression system itself can introduce new, and specific, product- and process-related impurities and substances that were not recognized in the production of the reference product. This is especially true if the reference product was made using a different system. For example, protein production in a mammalian-based cell line will need to control for different host cell proteins than a plant-based cell line, and products expressed in bacteria may have a greater likelihood for endotoxin contamination. The protein itself can be affected during its expression due to truncations, aggregates, and modifications.
It is important to note that not all impurities are detrimental to development. Levels should be based on ICH-guided drug substance and drug product impurity specifications and knowledge gained from the production process and batch history analysis. FDA requires a functionality and potency assessment. In many cases, as long as the safety and efficacy of the protein drug is maintained, the additional substances are not a concern. If the specific impurities are shown to impact functionality, further upstream and downstream process optimization may be required to obtain the necessary purity and potency levels. This can impact the product development plan from a safety and efficacy perspective as well.
Having an Integrated Drug Development Approach
CMC challenges related to upstream scale up and downstream purification strategies and the growing cost of product development have contributed to a shift in innovation and evaluation of product candidates in alternative systems and other technologies. Developers are considering new ways to expand product pipelines and will move to integrate those systems that provide them with a competitive advantage. The 351(k) pathway for biosimilars development will compel manufacturers to build quality into the process in the early stages, use a risk-based approach to proving comparability and characterization of their product, and to work together with key subject matter experts and the FDA to create a successful development program.
References
1. FDA. Quality Considerations in Demonstrating Biosmilarity to a Reference Listed Product (Draft Guidance). http://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/UCM291134.pdf
2. Walsh, G. (2010). Biopharmaceutical benchmarks 2010. Nature Biotechnology, 28 (9), 917-924.
About the Authors
Tom Fritz is the Managing Partner of Swiftwater Group. In his more than 20 years experience working in integrated drug development, he has managed company-wide drug and biologic programs and has been directly responsible for the success of numerous regulatory approvals.
Christine Lightcap, Ph.D., is a Senior Associate at Swiftwater Group who focuses on chemical and biological synthesis methodologies, driving all aspects of CMC planning and documentation. Christine earned a Ph.D. in biochemistry and molecular biology, with a concentration in structural biology and protein chemistry, from Thomas Jefferson University in Philadelphia.
Kundini Shah, M.S., is a Manager at Swiftwater Group with broad-based experience in early phase product development (small and large molecules). She supports several clients using alternative expression systems for the development of therapeutic proteins in the areas of project management, quality assurance, and regulatory affairs.