QbD for Better Method Validation and Transfer
More companies in the pharmaceutical industry today are adopting the principles of Quality by Design (QbD) for pharmaceutical development and manufacturing. Described in ICH Q8, Q9 and Q10 guidance documents, QbD enables enhanced process understanding, and a more systematic and scientific approach to development, so that better controls may be implemented. The end goal is more robust manufacturing processes than those that typically result from traditional approaches to drug development.
The QbD framework has many implications for manufacturers and regulators alike. This article describes how analytical methods can be viewed as "processes" and QbD concepts applied, to improve both method validation and transfer. Its goal is to stimulate thinking and discussion on how analytical method validation and transfer could evolve as industry increasingly adopts Quality by Design concepts.
But QbD cannot be considered without examining validation within a product lifecycle framework. The U.S. Food and Drug Administration (FDA) attempted to do just that in a recent draft guidance [1] designed to help achieve the goals of its “Pharmaceutical GMP's for the 21st Century – A Risk Based Approach” [2].
This guidance addresses some of the problems with traditional approaches to process validation, which were articulated by Moheb Nasr nearly five years ago [3]. Too narrow a focus on a "three-batch" approach to validation has encouraged a "don't rock the boat" mindset within the industry that can fail to foster continuous improvements in quality or efficiency.
The Need for Real World Verification
The same problems can be seen in analytical method validation and transfer, where the focus often shifts from ensuring the robustness of the method itself to ensuring the robustness of the documentation, and how well it will withstand regulatory scrutiny.
Methods for pharmaceutical products are normally validated by experts who have developed the method in accordance with ICHQ2 guidance (Validation of Analytical Procedures: Text and Methodology) or USP <1225> (Validation of Compendial Procedures). However, validation can be treated as a one-off event, with little consideration given to verifying how well the method will perform at everyday, "real world" operating conditions. All too often, regulators and industry use ICH Q2 or USP <1225> in a “check box” manner without considering the intent of these guidances, or the philosophy of method validation.

These issues continue after the method has been validated, when it is transferred to another laboratory. This transfer step requires that the knowledge of how to operate the method be transferred to those who will use it routinely, and that an exercise be performed, documenting that similar results have been obtained by both parties.
The transfer exercise is usually performed by the most competent analytical staff in the receiving laboratory, yet it may have little relationship with how the method will be used in routine, day to day operations.
It may come as little surprise, then, that most methods fail to perform as intended in the receiving laboratory, triggering efforts to identify variables that are causing the discrepancies, and repeated, costly testing and documentation.
The roots of method transfer failures can usually be traced to insufficient consideration of the routine operating environment during the method validation exercise, and lack of an effective process for capturing and transferring the tacit knowledge of the development analysts.
A QbD Framework for Analytical Method Life Cycle Management
If an analytical method is viewed, simply, as a process, whose output is data of acceptable quality, QbD concepts designed for manufacturing processes can be applied to analytical methods [4]. Thus, the concepts of lifecycle validation that are being developed for manufacturing processes might also be applied to analytical methods.
QbD is defined as “a systematic approach to development that begins with predefined objectives and emphasizes product and process understanding based on sound science and quality risk management” [5] and FDA [2] has proposed a definition for process validation that is “the collection and evaluation of data, from the process design stage throughout production, which establishes scientific evidence that a process is capable of consistently delivering quality products.”
When considering a lifecycle approach to method validation, a similar definition could be adopted “The collection and evaluation of data and knowledge from the method design stage throughout its lifecycle of use which establishes scientific evidence that a method is capable of consistently delivering quality data.”
From these definitions, it can be seen that there a number of key factors that are important in a QbD\Lifecycle approach. These include:
- The importance of having predefined objectives
- The need to understand the method, or being able to explain the method performance as a function of the method input variables
- The need to ensure that controls on method inputs are designed such that the method will deliver quality data consistently in all the intended environments in which it is used
- The need to evaluate method performance from the method design stage throughout its lifecycle of use.
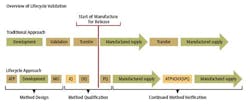
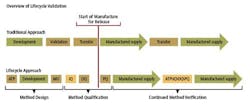
In alignment with the approach proposed in the draft FDA guidance for process validation, a three-stage approach could be taken with method validation. (See chart.)
Stage 1 – Method Design: Define method requirements and conditions and identify critical controls
Stage 2 – Method Qualification: Confirm that the method is capable of meeting its design intent.
Stage 3 – Continued Method Verification: Gain ongoing assurance to ensure that the method remains in a state of control during routine use.
Stage 1 - Method Design
The Method Design stage includes establishing the method performance requirements, developing a method that will meet these requirements and then performing appropriate studies to understand the critical method variables that must be controlled to assure the method is robust and rugged, The method design stage can be an iterative process which is repeated as required in accordance with the lifecycle phase of the product.
Method Performance Requirements
Utilising a QbD approach, it is essential at this stage that sufficient thought be given to the intended use of the method and that the objectives or performance requirements of the method be fully documented. This represents the Analytical Target Profile (ATP) [6] for the method.
To draw a parallel to qualification of new analytical equipment, the ATP for the method is effectively the equivalent of a User Requirement Specification that would be produced to support qualification of new analytical equipment (or the Design Qualification as defined in USP <1058>).
To build the ATP, it is necessary to determine the characteristics that will be indicators of method performance. Typically these characteristics will be a subset of those described in ICH Q2, such as accuracy or precision.
It is important, however, not to use the ICH Q2 characteristics in a check box manner but to consider the method's intended use. For example, the characteristics that are critical to the performance of an HPLC assay method intended to be used to quantify the active ingredient within a tablet might be quite different from those of an NIR method intended to be used to measure the end point of a blending operation. In the second case, accuracy may not be a critical method performance requirement in determining homogeneity.
Once the important method characteristics are identified, the next step is to define the target criteria- in other words, how accurate or precise the method should be. A key factor in choosing the appropriate criteria is the impact of method variation on the overall manufacturing process capability. Knowledge of the proposed specification limits and the expected process mean and variation is helpful in setting meaningful criteria.
Method Development
Once the ATP has been defined, an appropriate technique and method conditions must be selected in order to meet the requirements of the ATP. While method development is a very important part of the method lifecycle, it is not necessary to elaborate here since it has been extensively addressed in the literature.
Method Understanding
Based on an assessment of risk (i.e. the method complexity and the potential for robustness and ruggedness issues) one can perform an exercise focused on understanding the method to better understand what key input variables might have an impact on the method's performance characteristics. From this, one can identify a set of operational method controls.
Experiments can then be run to understand the functional relationship between method input variables and each of the method performance characteristics. Knowledge accumulated during the development and initial use of the method provides input into a risk assessment (using tools such as the Fishbone diagram and FMEA) which may be used to determine which variables need studying and which require controls.
Robustness experiments are typically performed on parametric variables using Design of Experiments (DoE) to ensure that maximum understanding is gained while minimizing the total number of experiments. Depending on the type of method, surrogate measures of characteristics such as accuracy or precision may be evaluated. An example might be peak resolution in a chromatographic method. It follows that well designed System Suitability requirements can be a valuable tool for assuring that a method is operating in a region where the ATP requirements will be met.
When developing an understanding of the method’s ruggedness, it is important to consider variables that the method is likely to encounter in routine use, such as in situations involving different analysts and different instruments. Tools such as measurement system analysis can be useful in providing a structured experimental approach to examining such variables.
Method Design Output
A set of method conditions will have been developed and defined which are expected to meet the ATP. Those conditions will have been optimized based on an understanding of their impact on method performance.
Stage 2 - Method Qualification
Once a set of operational method controls has been determined during the design phase, the next step is to qualify that the method will operate as intended. In a similar manner to equipment qualification, method qualification can be broken down into the stages of method installation qualification (MIQ), method operational qualification (MOQ), and method performance qualification (MPQ).
Method Installation
Qualification focuses on ensuring that the facility where the method will be used is prepared to use it. This process includes ensuring that the analytical equipment is qualified and that appropriate knowledge transfer and training of analysts has been performed.
The method conditions and detailed operating controls, along with all the knowledge and understanding generated during the design phase, must be conveyed to staff at the facility in which the method will be used. Performing a “method walkthrough” exercise with both the development and routine analytical teams can be extremely valuable in ensuring all tacit knowledge about the method is communicated and understood.
The extent of the IQ phase may vary depending on the level of pre-existing knowledge of the routine testing laboratory with the product, method or technique. For example, the laboratory that originally developed the method may merely ensure that other personnel who will use the method are trained.
The Method Operational Qualification phase is focused on demonstrating that the method meets its design intent. It is similar to “traditional method validation.” In the lifecycle approach, however, the focus is not about performing the traditional check box exercise to demonstrate compliance with ICH Q2. Instead, it is about demonstrating that the method meets the specific requirements that have been pre-defined in the Analytical Target Profile. A fundamental principle of this stage is that it should build on information that may have been generated in experiments already performed; for example, evidence of the selectivity or limit of quantification of a method may already exist from studies performed as part of method design.
The final stage in the qualification phase is to perform a MPQ exercise. At this stage, the actual manufactured supply samples are tested in facility, equipment and by personnel that will be used for routine analysis. The method should be operated exactly in accordance with the defined method controls and local operating procedures. As with the IQ stage above, the extent of the PQ exercise could vary, and would include for example confirming system suitability criteria are being routinely met.
Stage 3 - Continued Method Verification
The goal of this stage of the method lifecycle is to continually assure that the method remains in a state of control. This should include an ongoing program to collect and analyze data that relate to method performance---for instance, by setting criteria for replication of samples or standards during the analysis, by trending system suitability data or by trending the data from the regular analysis of a reference lot.
This activity aligns with the guidance in USP chapter 1010 on system performance verification. Close attention should also be given to any out of specification (OOS) or out of trend (OOT) results generated by the method once it is being operated in its routine environment. Ideally, using the new approach, laboratories should encounter fewer OOS results. If they do, it will be easier for staff to determine their root cause.
Change Control
During the lifecycle of a product, both the manufacturing process and the method are likely to experience a number of changes brought through unintentional deviations, continuous improvement activities or the need to operate the method and\or process in a different environment. It is essential that all changes to the method operating conditions be considered in light of the knowledge and understanding that exists on the method performance. If the method operating conditions are modified such they fall outside the known method operable region (determined as part of ‘method understanding’ during the original design phase) then the Method Qualification should be revisited. Likewise if the process changes and samples contain analytes at levels outside those covered in the method operational qualification exercise or have new interferences, a new ATP would need to be generated and a partial re-qualification exercise will be required to ensure the method is still capable of producing consistent and reliable results for the extended range or modified sample.
Where a method needs to be transferred to new location, appropriate Method Installation Qualification activities (including knowledge transfer) will need to be performed in addition to a Method Qualification exercise.
Consistent with QbD principles, the extent of requalification should be driven by scientific principles, as described previously
Other Scenarios
The approach to method qualification described above focuses on the activities that would typically be performed when a method is developed and used within a single company. Other scenarios exist where a laboratory may need to use a method for which it has no access to the original method design or qualification information such as in a contract testing laboratory. In these situations, it is important that an ATP is defined and documented. An appropriate qualification study is then performed to demonstrate that the method meets its ATP. This approach could also be applied to qualification of a pharmacopoeia method. By applying the lifecycle approach to pharmacopoeia methods it is envisaged that there would be no need for a separate USP chapter on method verification (USP <1226> Verification of Compendial Procedures).
Another scenario that needs to be considered is when a method is used to support drug development rather than drug manufacturing activities. In this case the method will still need to produce data that is scientifically sound - particularly as it may be used to release material for clinical supplies, support stability studies or support the development of process understanding. At this point in the product lifecycle however the focus is on understanding the method performance rather than demonstrating that it meets predefined acceptance criteria.
A key advantage of this approach for the above scenarios is the flexibility to perform a qualification against the specific ATP defined for the intended use of the method. This eliminates the current desire to create a qualification document against ICHQ2 in a check box manner which can lead to unnecessary headaches and additional work.
Since there is the potential for this approach to be adopted for all users of analytical methods, it also offers the potential to standardise the multiple, misrepresented terminology which currently exists within the industry. It would result in a harmonised method qualification approach with standardised terminology which would remove the current state of confusion between the terms; method validation transfer and verification and the many interpretations as to what is actually required for each.
In short, adopting a QbD approach to analytical method lifecycle management would have some significant implications for analytical scientists in the pharmaceutical industry. First, ICH Q2 and pertinent USP chapters would need to be revised to align with the lifecycle validation concepts described above. (The need for a revision of ICH Q2 as a consequence of increasing adoption of QbD concepts and use of PAT has also been identified [7].
The activities that were previously defined as “method transfer” (that is, knowledge transfer and confirmation of equivalence) would become a central component of the lifecycle validation approach (in other words, they would be described as method qualification activities) rather than being treated as distinct from validation.
Likewise, method validation as described in USP chapter <1225>, method verification as described in USP chapter 1226 and the proposed USP chapter on Transfer of Analytical Procedures would be encompassed within the lifecycle approach and it may be logical to combine those chapters.
The CMC section of regulatory submissions may also need to change. In a Quality by Design rather than “Quality by Testing” paradigm, it seems inappropriate that the validation details of the test methods should fill such a large proportion of a submission. With a lifecycle approach to method validation the submission would see an increased focus on the method design aspects and demonstration of the work performed to arrive at method understanding. This would parallel the increasing focus on process understanding in submission which has arisen since the introduction of ICH Q8.
Review of formal method qualification activity would become an inspection issue rather than a registration issue. Underpinning this approach is the need for effective lifecycle management of knowledge and understanding of the analytical method.
In summary, the switch to a QbD approach to method development is already beginning to bring improvements to the performance of analytical methods. It is now time to look to modernize and standardize industry’s approach to method validation and transfer. By aligning method validation concepts and terminology with those used for process and equipment qualification, there is an opportunity to both reduce confusion and complexity for analytical scientists as well as to ensure that efforts invested in method validation truly add value, rather than simply being a check box exercise.
|
Glossary: Method Development Method Understanding (MU) Method Installation Qualification (MIQ) Method Operational Qualification (MOQ) Method Performance Qualification (MPQ) |
About the Authors
Phil Nethercote is head of the Analytical Centre of Excellence in GSK’s manufacturing division, and chairman of the GSK Analytical Standards Group. Since joining GSK in 1987 he has held a number of roles associated with new product introduction. He has a degree in Chemistry from Heriott-Watt University and a PhD in HPLC from Stirling University.
Phil Borman is a manager in Analytical Sciences, Chemical Development within GSK and is currently accountable for the application of Statistics globally within his department. Prior to his current role, Phil worked in both Chemical and Pharmaceutical Development. He has an M.S. in chemistry from the University of Manchester and an M.S. in industrial data modeling from De Montfort University in Leicester.
Tony Bennett is Project Leader for Laboratory Support in Quality Analytical Sciences, Existing Product Support, Global Manufacturing Supply at GSK.
Gregory P. Martin is President of Complectors Consulting, LLC, in Pottstown, Pennsylvania.
Pauline McGregor is Analytical Method Support Specialist for PMcG Consulting of Oakville, Ontario. She has over 20 years experience in validation, R&D and quality systems in the pharmaceutical industry.
References
1. U.S. Food and Drug Administration (FDA). Draft Guidance for Industry
– Process Validation: General Principles and Practices. 2008.
2. FDA. “Pharmaceutical cGMPs for the 21st Century – A Risk-Based
Approach.” 2004.
3. Nasr, M. speaking at the 2005 AAPS Workshop on “Pharmaceutical
Quality Assessment - A Science and Risk-based CMC Approach
in the 21st Century.”
4. Borman, P., Nethercote, P., Chatfield, M., et al. Pharmaceutical
Technology, 31(10). 2007.
5. ICH Q8 (R2) – Pharmaceutical Development, November 2005.
6. Schweitzer, M., Pohl, M., Hanna-Brown, M., et al, Pharmaceutical
Technology, 34(2). 2010.
7. Ciurczak, E., ICH Guidances (Already) Show Their Age. www.
pharmamanufacturing.com/articles/2008/158.html